Int J Drug Res Clin. 2023;1:e2.
doi: 10.34172/ijdrc.2023.e2
Review Article
Pan Cyclin-Dependent Kinase Inhibitors for the Treatment of Breast Cancer
Sepideh Izadi 1, 2  , Mohammad Hojjat-Farsangi 3, Vahid Karpisheh 1, Farhad Jadidi-Niaragh 4, 5, *
, Mohammad Hojjat-Farsangi 3, Vahid Karpisheh 1, Farhad Jadidi-Niaragh 4, 5, *
Author information:
1Drug Applied Research Center, Tabriz University of Medical Sciences, Tabriz, Iran
2Student Research Committee, Tabriz University of Medical Sciences, Tabriz, Iran
3Bioclinicum, Department of Oncology-Pathology, Karolinska Institute, Stockholm, Sweden
4Immunology Research Center, Tabriz University of Medical Sciences, Tabriz, Iran
5Department of Immunology, Faculty of Medicine, Tabriz University of Medical Sciences, Tabriz, Iran
Abstract
Background:
Despite extensive attempts for the treatment of breast cancer (BC), it is the most prevalent cancer type among women, and its treatment remains elusive, particularly in patients with advanced disease. Although there are several therapeutic options, none of them is effective for complete relief, especially in metastatic patients. Cancer cells exhibit a high proliferation rate, which is usually associated with the dysregulation of cell cycle progress. Various proteins such as cyclin-dependent kinases (CDKs) and cyclins are involved in cell cycle modulation.
Methods:
Databases including PubMed, Scopus, WebofScience and Google Scholar were used to extract information. Articles published in English until 2022 were used.
Results:
Regarding the dysregulation of various CDKs in several cancer types, the pharmacologicalinhibitors of CDKs have extensively been evaluated to treat several cancer types such as BC. The blockade of CDKs strongly suppresses tumor growth through cell cycle arrest. Moreover, the combination of CDK inhibitors and other anti-cancer therapeutics has demonstrated potent synergistic effects on the treatment of various cancers.
Conclusion:
Therefore, various CDK inhibitors have been designed and evaluated as antiproliferative therapeutics to suppress the proliferation of cancer cells. Pan CDK inhibitors, including flavopiridol, dinaciclib, purvalanol A, SNS-032, and roscovitine, are the most effective CDK inhibitors investigated in several studies. They inhibit various CDKs such as the CDK1, 2, 4, 5, 6, 7, and 9. In this review, it is attempted to discuss the efficacy of Pan CDK inhibitors as an anti-cancer therapy in BC.
Keywords: CDK inhibitor, Cyclin-dependent kinases, Breast cancer
Introduction
Breast cancer (BC), the most prevalent women-related cancer, is characterized by the rapid and uncontrolled proliferation of mammary epithelial cells. It involves about 30% of new cancer diagnoses and 15% of cancer-triggered deaths globally. In addition, disease occurrence is significantly correlated with age.1 It should be noted that early diagnosis by advanced tools has potently improved survival time. Despite rigorous advances in identifying the immunopathogenesis of BC, the precise molecular mechanisms responsible for disease onset and progression remain elusive. Although several therapeutic approaches have been evaluated to treat BC, none of them was associated with complete remission, particularly in the advanced stages of the disease. Currently, chemotherapy, hormone therapy, surgery, and radiation therapy are the most common therapeutic approaches used to treat BC. Although cytotoxic drugs are an important part of the treatment regimen, the majority of patients with BC exhibit chemoresistance and cancer recurrence up to two years post-treatment. Therefore, the identification of novel efficient therapeutic approaches is a critical issue for the treatment of patients with BC. The precise identification of molecular mechanisms responsible for disease progression may help in the development of novel therapeutic approaches based on new molecular targets.2 Since cancer is characterized by the rapid and uncontrolled proliferation of malignant cells, it is assumed that molecules involved in cell cycle progress may be dysregulated during the tumorigenesis process. Cyclins and cyclin-dependent kinases (CDKs) are two critical types of molecules involved in cell cycle modulation in mammalian cells. The family of CDK proteins contains 13 members, including CDK1 to CDK13.3 They are classified into members responsible for the regulation of proliferation, including CDK1, 2, 3, 4, and 6, as well as those involved in the transcription process, including CDK7, 8, 9, 11, 12, and 13.4,5 Regarding the important role of these factors in the regulation of cancer cell proliferation, it is suggested that the blockade of these molecules can be considered a potent anti-cancer therapeutic approach in BC.6 Accordingly, several pharmacological CDK inhibitors such as CDK4/6 (e.g., palbociclib, ribociclib, and abemaciclib) and Pan CDK (e.g., flavopiridol, dinaciclib, purvalanol A, and roscovitine) inhibitors have been examined in several preclinical and clinical studies.7,8 Pan CDK inhibitors (i.e., flavopiridol, dinaciclib, purvalanol A, SNS-032, and roscovitine) are potent CDK inhibitors that can suppress various CDKs such as CDK1, CDK2, CDK5, CDK7, and CDK9.9 The pro-apoptotic and anti-proliferative effects of these CDK inhibitors are also demonstrated in BC cells, implying their potential as novel promising therapeutics alone or in combination with other anti-cancer drugs to treat BC patients.10 In this review, we intend to discuss studies performed to investigate the efficacy of Pan CDK inhibitors in the treatment of BC.
Breast Cancer
BC, the second cause of cancer-triggered deaths in women, is the most prevalent non-epidermal cancer which involves women worldwide.11 Patients may be afflicted with BC at different ages, but the average age of disease is 65 years in women. Intriguingly, men constitute about 1% of patients with BC.12 Despite several attempts to control the disease, an accumulating number of new cases is diagnosed each year.13 Due to advancements in early diagnosis and treatment strategies, the survival rate of BC patients is increased and mortality is decreased; nonetheless, high incidence and cancer recurrence are of the main challenges during the last three decades. The high prevalence of disease, particularly in developed countries, is partly due to increased risk factors, including physical inactivity, obesity, bad food behaviors, and modified reproductive patterns. It is also shown that an increased death rate after the age of 65 in women is related to modified fertility patterns.14 There are several risk factors affecting disease incidence; however, a few preventive strategies such as genetic screening and chemoprophylaxis by tamoxifen are available.15
Based on clinical and pathological characteristics, it is possible to classify patients with BC into five subtypes, including the HER2− enriched (HER2+, ER−, and PR−), luminal A (HER2−, PR+, ER+, and Ki67 < 14%), luminal B (HER2−/+, PR+, ER+, and Ki67 ≥ 14%), normal-like, and basal-like (HER2−, PR−, and ER−).16,17 The majority of estrogen receptor (ER)-α-expressing patients (about 70%) exhibit luminal A or B phenotype.18 The expression of this receptor is critical for disease progression, thus the blockade of its signaling by endocrine therapy has been proposed as an effective therapeutic strategy. However, resistance against endocrine therapy is the main challenge in this type of treatment.19 The expression of HER2 is also detected in nearly 25% of patients and is correlated with poor prognosis.20 Chemotherapy in association with radiation therapy is normally used to treat patients with advanced ER+ /Her2+ phenotype.21 Triple-negative BC (TNBC) constitutes 15% of BC patients and is the most progressive disease which is associated with high tumor size and tumor grade, cancer recurrence, and metastasis. Surgery and heavy chemotherapy are two available treatment options for these patients.22
Pharmacological CDK Inhibitors
Increased CDK activity potently enhances the proliferation of cancer cells, implying the use of CDK inhibitors as a novel promising anti-cancer therapeutic approach.23 Several CDK inhibitors have been designed and investigated in different cancer types, including non-selective (olomoucine, flavopiridol, kenpaullone, roscovitine, AT7519, AG-024322, SNS-032, and R547), selective (ryuvidine, fascaplysin, NU2058, purvalanol A, SU 9516, BML-259, P-276–00, and PD 0332991), and third-generation (dinaciclib and CR8) inhibitors. The majority of these drugs are entered into clinical trials, and some of them are also evaluated in patients with BC.24-26 This group of therapeutics exerts potent anti-proliferative effects on cancerous cells. The structure of CDK inhibitors is related to anion transporter polypeptides (ATP), and thereby they compete with kinases for binding to ATP.27 Selective inhibition and low toxicity have made CDK inhibitors novel and promising alternative therapeutics to current chemotherapies28 (Table 1).
Table 1.
CDK Inhibitors in Cancer Therapy
|
CDK Inhibitor
|
Administration
Route
|
IC50
|
Cancer Type
|
Clinical Trial Phase
|
| Palbociclib |
Oral |
CDK4: 11 nM,
CDK6: 16 nM |
BC and GI tumors |
III |
| Ribociclib |
Oral |
CDK4, 6: N/A |
GI cancers and BC |
III |
| Abemaciclib |
Oral |
CDK4, 6: N/A |
Lung cancer and BC |
I/(III) |
| Flavopiridol |
Intravenous |
CDK9: 10 nM
CDK7: 10 nM
CDK4: 20 nM
CDK1: 30 nM
CDK6: 60 nM
CDK2: 100 nM |
Prostate cancer, non-Hodgkin’s lymphoma, Renal cancer, colon cancer, and BC |
I/II |
| Dinaciclib |
Intravenous |
CDK5: 1 nM
CDK2: 1 nM
CDK1: 3 nM
CDK9: 4 nM |
BC |
III |
| Purvalanol A |
- |
CDC2/cyclin B:4 nM CDK2/cyclin E:35 nM
CDK2/cyclin A:70 nm
CDK5-p35:75 nM CDK4/cyclin D1:850 nM |
Breast cancer |
- |
| SNS-032 |
Intravenous |
CDK9: 4 nM
CDK2: 38 nM
CDK7: 62 nM |
BC, multiple myeloma, and chronic lymphocytic leukemia |
I |
| Roscovitine |
Oral |
CDK2: 0.1 μM
CDK7: 0.5 μM
CDK9: 0.8 μM
CDK1: 2.7 μM |
Advanced malignancies |
II |
Note. CDK: Cyclin-dependent kinase; IC50: The half maximal inhibitory concentration; GI: Gastrointestinal; BC: Breast cancer.
CDK4/6 Inhibitors
Palbociclib
Palbociclib, which is also known as PD0332991, Ibrance, commercialized by Pfizer; NY, USA, was developed as an oral selective CDK4/6 inhibitor for cancer therapy.29,30 It prevents the binding of ATP with CDK6/cyclin-D2, CDK4/cyclin-D1, and CDK4/cyclin-D3 complexes.31 Palbociclib was an efficient CDK inhibitor as evaluated in various preclinical studies on sarcomas, glioblastoma, neuroblastoma, bladder cancer, colorectal carcinoma, and gastric cancer.32-37 Moreover, ER+ and HER2+ patients with BC exhibited high responses to treatment with this drug.38,39 Moreover, retinoblastoma (Rb)-deficient BC patients had no response to treatment with this inhibitor29,40 because the CDK4/6-cyclin D complex promotes cell division through the phosphorylation of Rb. In fact, palbociclib prevents Rb phosphorylation by the blockade of CDK4/6. The combination therapy of patients with ER+ HER2- advanced BC in phase III clinical trial (PALOMA-2) by palbociclib and Letrozole led to the Food and Agriculture Organization (FDA) approval of this CDK inhibitor in March 2017.41
Ribociclib
Ribociclib(LEE-011, Kisqali developed by Novartis and Astex Pharmaceuticals) is a blocker of cyclin D1/CDK4 and CDK6 that inhibits Rb phosphorylation.42 The efficacy of this CDK inhibitor has been confirmed in the treatment of BC and other drug-resistant cancers.43 Based on positive results in clinical trials, ribociclib was approved by the US FDA in March 2017 for use in combination with aromatase inhibitors (AI), including fulvestrant or letrozole, to treat patients with ER+/HER2- advanced or metastatic BC. The administration of ribociclib to patients in a clinical trial could significantly improve progression-free survival in BC patients (25 months) compared to the placebo group (16 months).44-47
Abemaciclib
Abemaciclib, which is also known as LY2835219, trade names Verzenio and Verzenios developed by Eli Lilly, is a selective inhibitor of CDK4/6. Its affinity to CDK4 is significantly (14 times) higher than CDK6.48 This CDK inhibitor can also block the function of several other kinases at low concentrations (lower than 100 nM).49 Although ribociclib and palbociclib are structurally based on the pyrido [2,3-d] pyrimidin-7-one scaffold, the structure of abemaciclib is related to the 2-anilino2,4-pyrimidine-[5-benzimidazole].49 Several preclinical studies on various cancer models such as glioblastoma, head, and neck squamous cell carcinoma, melanoma, and BC have shown the anti-cancer activity of this drug.50-53 The anti-cancer potential of abemaciclib has been demonstrated in various cancer types such as bladder cancer, melanoma, and BC.37,50,54 Regarding these anti-tumor functions, it was considered a breakthrough therapy for BC by the FDA in October 2015 and was approved for the treatment of hormone receptor-positive, HER2 - advanced, or metastatic BC on September 28, 2017.
Pan CDK Inhibitors
Flavopiridol (Alvocidib)
Flavopiridol or alvocidib (also known as L86-8275 or NSC 649890) is a semisynthetic flavonoid alkaloid and inhibits CDK9. The drug is a semisynthetic analog of rohitukine extracted from Amoora rohituka and Dysoxylum binectariferum. The initial evaluation of flavopiridol showed its anti-cancer potential in 60 different human cancer cell lines, representing its high and non-tumor type-specific function.55 It could block CDK9 activity, suppress transcription, induce apoptosis, and prevent angiogenesis.56 Flavopiridol suppresses the function of positive transcription elongation factor b (P-TEFb) which leads to the blockade of mRNA production.57 This CDK inhibitor is currently under clinical evaluation by Tolero Pharmaceuticals, Inc for the treatment of acute myeloid leukemia. Flavopiridol is also evaluated for the treatment of atherosclerotic plaque formation and arthritis.58 Recently, the FDA has donated an orphan drug topic for flavopiridol for the treatment of patients with acute myeloid leukemia; however, little is known regarding its efficacy for the treatment of BC, and this issue needs to be further investigated in future studies.
Dinaciclib
Dinaciclib, also known as SCH727965 or MK-7965, is another potent CDK inhibitor, which interacts with the acetyl-lysine recognition site of bromodomains and blocks the function of various CDKs such as CDK1, 2, 5, and 9. Dinaciclib could significantly induce apoptosis in cancer cells in vitro and suppresses tumor growth in both tumor-bearing animal models and human cancers.59-62 Accordingly, dinaciclib could suppress the progression of melanoma in a p53-dependent manner.63 It also prevented the expansion of chronic lymphocytic leukemia64 and osteosarcoma cells65 through the induction of apoptosis. Anti-cancer effects were also observed in the murine xenograft models of pancreatic cancer.66 The anti-cancer activity of this drug has also been demonstrated in BC cell lines and xenograft BC models.61,62,67 In vitro studies indicated the high activity of dinaciclib in TNBC.68 Dinaciclib also showed higher activity and lower adverse effects compared to flavopiridol in clinical trials.59 Potent anti-cancer function observed in preclinical studies led to the entrance of dinaciclib into the initial phases of clinical trials in various cancer types such as BC.69-71 It is now under consideration in clinical trials for the treatment of various cancer types, including advanced BC (phase II),72 non-small cell lung cancer (NSCLC, phase II),73 multiple myeloma (phase II), advanced melanoma (phase II), and chronic lymphocytic leukemia (phase III). It was developed by Merck et al and assigned as an orphan drug by the FDA.74
Purvalanol A
Purvalanol A is another CDK inhibitor that selectively suppresses CDK1 and CDK2 and arrests the cell cycle at G1/M and G2/S phases.75 Purvalanol A has shown synergism with chemotherapeutics such as paclitaxel to suppress the growth of Hela cells in part through reducing the expression of Bcl-2 anti-apoptotic proteins.76-78 The anti-cancer function of this CDK inhibitor is demonstrated in vitro by inducing apoptosis in MDA-MB-231 and MCF-7 BC cell lines.75
SNS-032
SNS-032, also known as BMS 387032 and developed by Sunesis, has a thiazole unit and selectively inhibits CDK2, 7, and 9.79 The comparison of this CDK inhibitor with flavopiridol represented its lower toxicity, higher suppression of transcription, and higher apoptosis induction.80 Treatment of BC cells such as MDA-MB-435, MCF-7, and MDA-MB-435 led to the apoptosis of cancer cells partly through the downregulation of Mcl-1 and XIAP proteins. Moreover, the administration of this CDK inhibitor into MDA-MB-435 nude murine xenografts was associated with tumor regression, implying the efficacy of SNS032 in the treatment of BC.81
Roscovitine
Roscovitine, also known as seliciclib, CYC202, and developed by Cyclacel, was first generated by De Azevedo et al as a potent member of CDK inhibitors.82,83 The oxidative reaction of the hydroxymethyl group of the C2 amino alcohol at the purine core led to the generation of this compound, which is soluble in DMSO and HCl (up to 50 mM).84 Rapid metabolism of roscovitine by CYP2B6 and CYP3A4 leads to the generation of carboxylate PMF30-128 and M1/M2 glucuronides.85 This purine analog can potently suppress various CDKs such as CDK1, 2, 5, 7, and 9 (with an IC50 of 0.2-0.7 μM), whereas affects the function of CDK4, 6, and 8 (with an IC50 of higher than 100 μM). It can also inhibit several kinases, including IRAK4, FAK, ERK2, ERK1, EPHB2, DYRK1A, CK1δ, CK1α, and CaMK2 in the concentrations of 1 to 40 μm.82,86,87 It is indicated that roscovitine can arrest cell cycle progression at all stages, including G0, G1, S, or G2/M, which relies on cancer type, treatment time, and administrated dose. This wide spectrum impact on the cell cycle can disrupt several signaling pathways, including JAK-STAT,88 ER,89 p53,90 NF-κβ,91 and Ras-MAPK.92 Consequently, it can reduce the expression of several genes involved in the survival of cancer cells (e.g., XIAP,93 Mcl-1,94 survivin,93 and Bcl-295) and increase Bcl-x,96 p53,97 and p53AIP1.98 The pro-apoptotic effects of this CDK inhibitor have synergistic impacts when it is combined with chemotherapeutics.99-102 Accordingly, its synergism has been shown with several chemotherapeutics and anti-cancer drugs such as doxorubicin, tamoxifen, etoposide, irinotecan, cisplatin, trastuzumab, alemtuzumab, vinblastine, 5-fluorouracil, and paclitaxel.100,103 It has been reported that roscovitine exerts potent anti-tumor activity partly through the upregulation of the pro-apoptotic Bcl-x protein. Roscovitine also suppresses the expansion of various cancer cells such as MCF-7 BC, lung cancer, HeLa cervical cancer, and HCT116 colon cancer cells through cell cycle arrest at G1/S and G2/M phases. It is proposed that roscovitine can be an effective anti-cancer therapeutic for hormonal therapy-sensitive patients with BC. Moreover, the anti-proliferative effects of roscovitine are through decreasing the expression of cyclin D1 via the MAPK pathway and depend on ER-α expression status.92,104 Interestingly, roscovitine also enhances the autophagy process.87,95,97
In addition to cancer, the efficacy of roscovitine as a strong CDK/cyclin E inhibitor has been confirmed in various pathologic conditions such as glomerulonephritis, viral infections, and neurodegenerative disorders. Since it can induce apoptosis in neutrophils, it is proposed that roscovitine could be used as a potential treatment for chronic inflammatory diseases.105 As an anti-cancer therapeutic, it was effective in several cancer cell lines in vitro. Furthermore, several preclinical studies have confirmed its anti-cancer potential in various cancer models. For instance, the intraperitoneal administration of roscovitine into LoVo human colorectal cancer-bearing CD1 nude mice led to a significant reduction in the tumor size. Similar results were observed following the oral administration of roscovitine into the tumor xenografts of MESSA-DX5 human uterine carcinoma cells.106 Orally treated HCT116 human colon tumor-bearing nude mice with roscovitine demonstrated significantly smaller tumors compared to control mice.107 Intraperitoneal administered roscovitine also potently arrested tumor growth in the A4573 Ewing’s sarcoma and PC-3 prostate cancer xenografts.108,109 Similar anti-tumor functions were detected in the osteosarcoma tumor xenograft in B6D2F1 mice110 and HT29 human colon cancer xenografts in nude mice111 following treatment with roscovitine. Additionally, the treatment of RAMOS or HBL-2 tumor-bearing immunodeficient mice with roscovitine led to potent tumor regression.112 Likewise, other preclinical studies performed in BC animal models will be discussed in the next section.113,114
Roscovitine is now under clinical evaluation for the treatment of various cancers. Investigating the efficacy of this drug in patients with NSCLC showed some adverse effects such as vomiting, nausea, transient hypokalemia, and transient elevations in serum creatinine and liver function. Its assessment in patients with B-cell lymphomas was associated with the downregulation of MCL1 protein.94 Roscovitine is also used in phase I trial against glomerulonephritis and phase II trials against lung cancer and BC.115
Pan CDK Inhibitors in Breast Cancer
Flavopiridol
This section reviews studies on the effectiveness of this CDK inhibitor in the treatment of BC. Schwartz et al found that flavopiridol enhances the cellular toxicity of mitomycin-C (MMC), a chemotherapeutic agent, by stimulating drug-induced apoptosis in MDA-MB-468 and KN-74 tumor cell lines. Apoptosis enhancement caused by flavopiridol may somewhat depend on PKC and is not associated with a specific area of the cell cycle.116 Furthermore, Motwani et al examined events related to apoptosis and the cell cycle associated with the combined treatment of MKN-74 and MCF-7 cell lines with paclitaxel and flavopiridol. They reported that flavopiridol enhances apoptosis caused by paclitaxel via increasing the activation of caspases and poly (ADP-ribose) polymerase (PARP) cleavage. The cell treatment with flavopiridol inactivates Cdc-2 kinase, which inhibits paclitaxel mitotic arrest.117 Flavopiridol also inhibits the growth of MDA-MB-435 and 435.eB cells; regardless of excessive c-erbB-2 expression, it causes apoptosis and suppresses the expression of MMPs and invasion of BC cells.118 Carlson et al concluded that the flavopiridol-induced reduction in cyclin D1 due to the suppression of cyclin D1 promoter transcription is a specific primary event in MCF-7 and U2OS human cell lines.119 Similarly, Nahta et al that the activity of CDK4/6 is particularly essential for erbB2-mediated transformation and mutually regulates erbB2 expression. They reduced the level of erbB2 and suppressed the cell proliferation using the combination therapy of (erbB2 + ) BC cells with flavopiridol and herceptin against cyclin D1 and erbB2.120 In another study, they showed that the expression of epidermal growth factor receptor (EGFR) and erbB2 proteins in BT474, MDA-MB-453, and SKBR was rapidly downregulated after the combination therapy of trastuzumab and flavopiridol.121 It has also been shown that the combination of flavopiridol and trastuzumab synergistically inhibits the Ras-MAPK and/or PI3K/Akt pathway and reduces the levels of Cdks and/or cyclin D1, as well as the S-phase transition in the HBL-100, MDA-MB-231/453, and MCF7.122 Wall et al found that Thr34 phosphorylation is necessary to retain the expression of survivin in tumor cells, and p34cdc2 kinase inhibition in the arrested cells of the mitotic phase using flavopiridol leads to the downregulation of survivin and the increased apoptosis in MCF-7 and HeLa cells.123 Further, the treatment of SKBR-3 and MB-468 with flavopiridol and epothilone B altered the Bax structure and greatly reduced Bcl-xL, cIAP-2, XIAP, and Mcl-1 expression, and increased apoptosis. However, MB-468/Bcl-2 cells were less sensitive to Epo B and FP.124 It has also been represented that flavopiridol reduces the expression of the α5β1 integrins and the adherence to fibronectin, inhibits Akt phosphorylation induced by fibroblast growth factor 2 (FGF-2), and leads to the survival of T-47D and MCF-7 cells.125 According to the report of another study, the combined therapy of MCF-7 and MDA-MB-231 cells using flavopiridol and gemcitabine resulted in apoptosis induction, cell growth inhibition, and cell accumulation in the S phase.126 Meyer et al evaluated the effect of flavopiridol on germ cell tumors-derived cell lines 2102 EP, NCCIT, and NT2 compared to cell lines derived from MCF7, HeLa, and SKOV. They found that GCT cells were more sensitive to flavopiridol than the other cell lines. Depending on the correlation of RB and E2F1 transcription factor, flavopiridol induces apoptosis or cell cycle arrest.127 Another study represented that flavopiridol promotes the degradation of the c-FLIP anti-apoptotic protein, abrogates the NF-kβ activity, downregulates the XIAP, induces TRAIL signaling, and increases the activation of caspase-8 and apoptosis in breast tumor cells.128 Flavopiridol decreases proapoptotic proteins (Bax, Bak, and PUMA-α) and FLIPL expression and promotes TRAIL-induced apoptosis in BC and myeloma cancer cells. Moreover, the knockdown of the FLIPL gene with small interfering RNA (siRNA) sensitizes MM1S cells to TRAIL.129 Data from another study confirmed that flavopiridol in combination with vorinostat inhibits AKT and ERK1/2 function, decreases the expression of various inhibitors of internal and external apoptosis pathways (XIAP, BCL-xL, and c-FLIP-l/s), activates cathepsin-dependent protease pathways, and leads to increased apoptosis in BC cells.130 Fornier et al concluded that flavopiridol enhances the docetaxel-dependent apoptosis in patients with ovarian, breast, and pancreatic carcinoma in a time- and sequence-dependent manner. To induce this effect, docetaxel should be administrated at least 4 hours before flavopiridol in vivo.131 It was also represented that the suppression of CDK4/6 and cyclin D1 by flavopiridol leads to decreased or increased stem-like cell function and migration in (ER+) and (ER-) BC cells, respectively.132 In addition, the simultaneous targeting of Ras-MAPK pathways and CDK with sorafenib (SFN) and flavopiridol enhances the cellular toxicity and lethality induced by SFN in the cell lines with the KRAS-BRAF mutation or EGFR/HER-2 overexpression, respectively. Furthermore, the combination of flavopiridol and SFN inhibits the Mcl-1, Rb-E2F signaling, Erk, tumor growth, and pulmonary metastatic burden in vivo.133 Hicks et al found that JUNB plays an important role in the survival of BC cells treated with kinase inhibitors. Thus, they showed that FP treatment initially lowers Pol IIser2p levels, as well as MCL1, JUNB, MYC, and CEBPD expression, in MDA-MB-231/468, and SKBR3 BC cells.134 Brenner et al revealed that organic anion-transporting polypeptides (OATP1B1/B3 and OATP2B1) play a substantial role in the uptake of flavopiridol to cancer cells. They indicated that the use of OATP inhibitors such as rifampicin inhibits the flavopiridol uptake in OATP1B3/B1- and OATP2B1-transfected CHO cells. Moreover, after flavopiridol treatment, wild-type ZR-75-1 cells were blocked in the G1 phase and G2/M, while OATP1B1 knockdown ZR-75-1 cells were only inhibited in the G1 stage.135 Another study reported that the treatment of MCF-7 cells with flavopiridol induces oxidative stress, alters multiple metabolic pathways, including glutathione metabolism, glycerophospholipid metabolism, and polyamine metabolism, and finally leads to cell death. Additionally, spermine and spermidine, which exacerbate cancer progression, are downregulated after treatment due to the G1 phase arrest.136 Other studies represented that autophagy increases the survival of cancer cells after radiation and chemotherapy. Accordingly, the findings of Okada et al revealed that treatment with flavopiridol/CDK4 inhibitor, along with autophagy inhibition using chloroquine or BECN1/ATG5 knockdown, inhibits stress-induced autophagy, arrests the cell cycle, and causes apoptosis in MDA-MB435S, BT474, SW480, A431, and SKBr3 cells.137 It has also been shown that the treatment of MCF-7 cells with flavopiridol reduces the expression of anti-apoptotic proteins in the BCL-xl and IAP family, and increases the expression of BAX pro-apoptotic proteins independent of the ER status.138 Flavopiridol also inhibits the growth of CD44+/CD24- BC stem cells, ribosome biogenesis, and translation139 (Table 2, Figure 1).
Table 2.
Studies Related to the Role of Flavopiridol in Breast Cancer
|
Cell Lines
|
Drug/SiRNA
|
Human/Animal Model
|
In vivo/
In vitro
|
Study Result
|
Reference
|
MKN-74
MDA-MB-468 |
Flavopiridol, MMC |
- |
In vitro |
Flavopiridol and MMC induce apoptosis in MDA-MB-468 and MKN-74 cells |
116
|
MKN-74
MCF-7 |
Flavopiridol, Paclitaxel (Taxol) |
- |
In vitro |
Flavopiridol induces caspase activation and apoptosis in gastric and BC cells treated with Paclitaxel |
117
|
MDA-MB-435
435.eB1
435.eB4 |
Flavopiridol |
- |
In vitro |
Flavopiridol inhibits the MMPs expression, growth, and metastasis of BC cells |
118
|
MCF-7
U2OS |
Flavopiridol |
- |
In vitro |
Flavopiridol leads to a fast decrease in cyclin D1 levels |
119
|
NIH3T3 MCF-7 SKBR3
MDA-MB-453 |
Flavopiridol, trastuzumab (Herceptin) |
- |
In vitro |
Combination therapy of (erbB2+) BC cells with flavopiridol and trastuzumab reduces erbB2 levels and suppresses cell proliferation |
120
|
MDA-MB-453
BT474
SKBR3 |
Flavopiridol, trastuzumab, Z-VAD-FMK (Caspase inhibitor), AG99, AG1478 |
- |
In vitro |
The combination of flavopiridol and trastuzumab reduces EGFR and erbB2 expression |
121
|
MCF7
MDA-MB-453
MDA-MB-231
HBL-100 |
Flavopiridol, trastuzumab, PD98059 (MEK1 inhibitor), LY 294002 (PI3K inhibitor) |
- |
In vitro |
Flavopiridol and trastuzumab inhibit the PI3K/Akt and/or Ras-MAPK pathway, DNA synthesis, and cell proliferation |
122
|
MCF-7
HeLa |
Flavopiridol |
- |
In vitro |
Inhibition of p34cdc2 kinase in arrested cells using Flavopiridol leads to decreased Survivin levels and increased apoptosis in MCF-7 and HeLa cells |
123
|
| MB-468 SKBR-3 |
Flavopiridol, Taxotere (docetaxel), Epothilone B (Epo B), Proteosome inhibitor ALLnL |
- |
In vitro |
FP reduces the IAP and Bcl-2 family levels, alters the BAX structure, and induces apoptosis in BC cells |
124
|
MCF-7
T-47D |
Flavopiridol, MEK inhibitors: UO126,
PD98059, P38 inhibitors: SB203580, D169316 |
- |
In vitro |
Flavopiridol suppresses the expression of α2, α5, β3, and β4 integrins upregulated by FGF-2 and declines the survival of BC cells |
125
|
| MCF-7 (ER+) MDAMB-231 (ER-) |
Flavopiridol, gemcitabine |
- |
In vitro |
Flavopiridol and gemcitabine lead to apoptosis by upregulating P16 and p21WAF1 cell cycle inhibitors |
126
|
NTera2, 2102 EP NCCIT
MCF-7
SKOV, HeLa |
Flavopiridol, cisplatin, paclitaxel |
- |
In vitro |
Flavopiridol induces cell death in cancer cells, especially in GCT cell lines |
127
|
MCF-7, MDA-MB-231, EVSA-T Jurkat, MDAMB-468
SKBR-3, and BT474
MDA-MB-435S |
Flavopiridol, proteasome inhibitor: MG-132,
caspases inhibitor: Z-VAD-FMK, recombinant human TRAIL (residues 95-281) |
- |
In vitro |
Flavopiridol induces TRAIL signaling and apoptosis and promotes the degradation of c-FLIP anti-apoptotic protein in BC cells |
128
|
MM1S
MDA-MB-468 |
Flavopiridol, c-FLIP-siRNA |
- |
In vitro |
Treatment with flavopiridol through suppressing transcription and reducing FLIPL expression causes the apoptosis of breast and myeloma cancer cells |
129
|
| MDAMB-231 MCF7 |
Flavopiridol, vorinostat |
- |
In vitro |
Flavopiridol in combination with vorinostat inhibits AKT and ERK1/2 functio\n, minimizes the expression of XIAP, BCL-xL, and c-FLIP-l/s anti-apoptotic proteins, and leads to increased apoptosis in BC cells |
130
|
| - |
Flavopiridol, focetaxel |
Human |
In vivo |
Flavopiridol enhances the docetaxel–dependent apoptosis in patients with ovarian, breast, and pancreatic carcinoma in a time- and sequence-dependent manner |
131
|
MDA-MB-231
MDA-MB-468 (ER-)
MCF7, T47D (ER+) |
Flavopiridol, PD0332991 |
- |
In vitro |
Flavopiridol leads to a decrease or an increase in the migration in (ER+) and (ER-) BC cells, respectively |
132
|
MDA-MB-468,
MDA-MB-231 SK-BR3, MCF-7 |
Flavopiridol (FPD), sorafenib (SFN)
lapatinib, erlotinib, U0126, LY294002, U73122, and AZD5438 PD0332991 |
BalbC-RAG2−/− |IL2Rγc−/− mice |
In vivo
In vitro |
FPD/SFN combinations cause the inhibition of tumor growth and the reduction of pulmonary metastatic burden in mutant RAS/RAF and (EGFR/HER2-+) BC model |
133
|
MDA-MB-231
MDA-MB-468
MCF10Amet
SKBR3 |
Flavopiridol, JUNB siRNA, Actinomycin D, U0126 (MEK1 inhibitor), sorafenib, doxorubicin |
- |
In vitro |
FPV treatment is used to delay the expression of the JUNB gene in BC cells |
134
|
ZR-75-1
CHO |
Flavopiridol, OATP inhibitors: bromsulphophthalein (BSP), and rifampicin |
- |
In vitro |
OATP1B1/B3 and OATP2B1 act as transporters for the cellular uptake of flavopiridol, and the use of OATP inhibitors rifampicin inhibits the flavopiridol uptake in OATP1B3/B1- and OATP2B1-transfected CHO cells |
135
|
| MCF-7 |
Flavopiridol |
- |
In vitro |
Treatment of MCF-7 cells with Flavopiridol induces the G1 phase arrest and oxidative stress, as well as lipid oxidation and mitochondrial lesions, and finally leads to cell death |
136
|
SW480, A431
KMST-6
NCI-N87
BT474
MDA-MB231
MDA-MB435S
SKBr3
MCF7 |
Flavopiridol
CDK4 inhibitor: 2-bromo-12 and 13-dihydro-5 H-indolo-dione [2 and 3-a] pyrrolo [3 and 4-c] carbazole-5 and 7 (6H)-dione
Autophagy inhibitior: Chloroquine (CQ), ATG5 siRNA, BECN1 siRNA |
- |
In vitro |
Flavopiridol, or CDK4 inhibitor, along with autophagy inhibition inhibited stress-induced autophagy, arrests the cell cycle, and causes apoptosis in some cell lines |
137
|
| MCF-7 |
Flavopiridol (FP) ATG5 siRNA |
- |
In vitro |
Treatment of MCF-7 cells with Flavopiridol reduces the expression of the BCL-xl and IAP family, while it increases the BAX expression |
138
|
| MCF7 (CD44+/CD24-) |
Flavopiridol |
- |
In vitro |
Flavopiridol inhibits the growth of CD44+/CD24- BC stem cells |
139
|
Note. SiRNA: Small interfering RNA; MMC: Mitomycin-C; FGF: Fibroblast growth factor; BC: Breast cancer; ER: Estrogen receptor; TRAIL: Tumor necrosis factor-related apoptosis-inducing ligand; c-FLIP: Cellular FLICE (FADD-like IL-1β-converting enzyme)-inhibitory protein; CDK: Cyclin-dependent kinase; GCT: Giant cell tumor; FPV: Feline panleukopenia virus; OATP: Organic anion transporter polypeptide; CHO: Chinese Hamster Ovary; EGFR: Epidermal growth factor receptor.
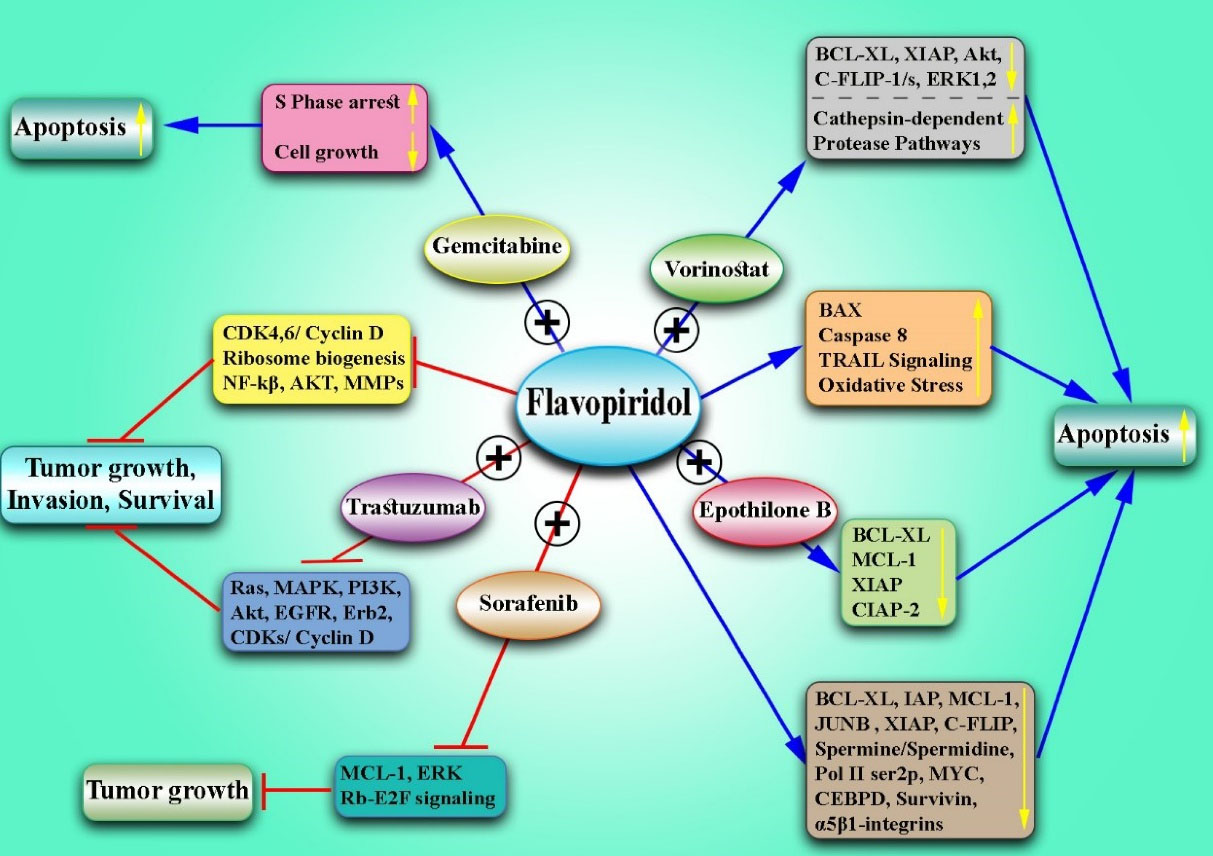
Figure 1.
The Effects of Flavopiridol Alone and in Combination With Other Drugs for the Treatment of Breast Cancer. Note.The data illustrate that Flavopiridol inhibits the growth of CD44 + /CD24 - BC stem cells, ribosome biogenesis, and translation. Moreover, it reduces the expression of the BCL-xl and IAP family, increases the expression of BAX pro-apoptotic proteins, down-regulates the spermine and spermidine, induces oxidative stress, lowers Pol IIser2p levels and MCL1, JUNB, MYC, and CEBPD expression, and suppresses the CDK4/6 and cyclin D1. Flavopiridol decreases proapoptotic proteins (e.g., Bax, Bak, and PUMA-α) and promotes TRAIL-induced apoptosis. Flavopiridol promotes the degradation of the c-FLIP anti-apoptotic protein, abrogates the NF-kβ activity, down-regulates XIAP, induces TRAIL signaling, and increases the activation of caspase-8 and apoptosis. In addition, Flavopiridol declines the expression of the α5β1 integrins and the adherence to fibronectin and inhibits Akt phosphorylation induced by FGF-2, and survival. It also leads to a drop in survivin surface and inhibits the MMP expression and invasion in the BC cells. The combination of Flavopiridol and Sorafenib inhibits the Mcl-1, Rb-E2F signaling (Erk), and tumor growth. Moreover, Flavopiridol in combination with vorinostat inhibits AKT and ERK1/2 function, decreases the expression of XIAP, BCL-xL, and c-FLIP-l/s, activates cathepsin-dependent protease pathways, and leads to increased apoptosis in BC. Flavopiridol and Gemcitabine resulted in apoptosis induction, cell growth inhibition, and cell accumulation in the S phase. Flavopiridol and Epothilone B altered Bax’s structure and greatly reduced Bcl-xL, cIAP-2, XIAP, and Mcl-1 expression and increased apoptosis. The combination of Flavopiridol and trastuzumab synergistically inhibits the Ras-MAPK and/or PI3K/Akt pathway, reducing the levels of EGFR, erbB2, Cdks, and cyclin D1. BC: Breast cancer; NF-kβ: Nuclear factor kappa β; XIAP: X-linked inhibitor of apoptosis protein; Bcl-xl: B-cell lymphoma-extra-large; IAP: Inhibitors of apoptosis proteins; BAX: Bcl-2-associated X protein; MCL1: Myeloid cell leukemia 1; CEBPD: CCAAT Enhancer binding protein delta; BAK: Bcl-2 homologous antagonist/killer, PUMA: p53 upregulated modulator of apoptosis; TRAIL: TNF-related apoptosis-inducing ligand; c-FLIP: FLICE-like inhibitory protein; NF-κβ: Nuclear factor kappa light chain enhancer of activated B cells; XIAP: X-linked inhibitor of apoptosis protein; FGF-2: Basic fibroblast growth factor (bFGF); MMP: Matrix metallopeptidase; cIAP-2: Cellular inhibitor of apoptosis 1; PI3K: Phosphoinositide 3-kinases; EGFR: Epidermal growth factor receptor.
.
The Effects of Flavopiridol Alone and in Combination With Other Drugs for the Treatment of Breast Cancer. Note.The data illustrate that Flavopiridol inhibits the growth of CD44 + /CD24 - BC stem cells, ribosome biogenesis, and translation. Moreover, it reduces the expression of the BCL-xl and IAP family, increases the expression of BAX pro-apoptotic proteins, down-regulates the spermine and spermidine, induces oxidative stress, lowers Pol IIser2p levels and MCL1, JUNB, MYC, and CEBPD expression, and suppresses the CDK4/6 and cyclin D1. Flavopiridol decreases proapoptotic proteins (e.g., Bax, Bak, and PUMA-α) and promotes TRAIL-induced apoptosis. Flavopiridol promotes the degradation of the c-FLIP anti-apoptotic protein, abrogates the NF-kβ activity, down-regulates XIAP, induces TRAIL signaling, and increases the activation of caspase-8 and apoptosis. In addition, Flavopiridol declines the expression of the α5β1 integrins and the adherence to fibronectin and inhibits Akt phosphorylation induced by FGF-2, and survival. It also leads to a drop in survivin surface and inhibits the MMP expression and invasion in the BC cells. The combination of Flavopiridol and Sorafenib inhibits the Mcl-1, Rb-E2F signaling (Erk), and tumor growth. Moreover, Flavopiridol in combination with vorinostat inhibits AKT and ERK1/2 function, decreases the expression of XIAP, BCL-xL, and c-FLIP-l/s, activates cathepsin-dependent protease pathways, and leads to increased apoptosis in BC. Flavopiridol and Gemcitabine resulted in apoptosis induction, cell growth inhibition, and cell accumulation in the S phase. Flavopiridol and Epothilone B altered Bax’s structure and greatly reduced Bcl-xL, cIAP-2, XIAP, and Mcl-1 expression and increased apoptosis. The combination of Flavopiridol and trastuzumab synergistically inhibits the Ras-MAPK and/or PI3K/Akt pathway, reducing the levels of EGFR, erbB2, Cdks, and cyclin D1. BC: Breast cancer; NF-kβ: Nuclear factor kappa β; XIAP: X-linked inhibitor of apoptosis protein; Bcl-xl: B-cell lymphoma-extra-large; IAP: Inhibitors of apoptosis proteins; BAX: Bcl-2-associated X protein; MCL1: Myeloid cell leukemia 1; CEBPD: CCAAT Enhancer binding protein delta; BAK: Bcl-2 homologous antagonist/killer, PUMA: p53 upregulated modulator of apoptosis; TRAIL: TNF-related apoptosis-inducing ligand; c-FLIP: FLICE-like inhibitory protein; NF-κβ: Nuclear factor kappa light chain enhancer of activated B cells; XIAP: X-linked inhibitor of apoptosis protein; FGF-2: Basic fibroblast growth factor (bFGF); MMP: Matrix metallopeptidase; cIAP-2: Cellular inhibitor of apoptosis 1; PI3K: Phosphoinositide 3-kinases; EGFR: Epidermal growth factor receptor.
Dinaciclib
Studies on BC treatment with this inhibitor will be discussed in this section. Mita et al evaluated the safety and efficacy of dinaciclib, compared with capecitabine, in patients with advanced BC who have previously undergone treatment. They demonstrated that dinaciclib treatment has a poor antitumor function as capecitabine, with 1 verified partial response (PR), 1 unverified PR, an overall response rate of 8%, and a time to disease progression lower than capecitabine treatment in patients with (ER+/HER2-) BC. Additionally, dinaciclib therapy has anti-tumor activity and is commonly safe and tolerable, but its efficacy is less than capecitabine.72 The findings of another study represented that the combination of dinaciclib and epirubicin is not an effective treatment selection for TNBC and is associated with considerable toxicity.140 It has also been reported that CDK9, cyclin B1, and MYC can be used as potential therapeutic targets in TNBC. Dinaciclib decreases cyclin B1 and MYC expression through CDK9 inhibition, arrests the G2/M phase of the cell cycle, and induces apoptosis in TNBC patient-derived xenograft (PDX) both in vitro and in vivo. Furthermore, CDK1/cyclin B1 knockdown using siRNA in MDA-MB-231 and BT549 cells greatly inhibits cell proliferation and induces apoptosis. In addition, the knockdown of the P-TEFb subunit of CDK9 by siRNA leads to decreased levels of MYC and cyclin B1 in TNBC cell lines.68 Recent studies showed that ER phosphorylation at the S294 site (pS294) is essential for the transcription of the ER-dependent gene and is mediated by an unknown CDK. Scott et al also reported that CDK2 is involved in the initial formation of pS294 as a mediator, and the use of dinaciclib, a selective CDK2 inhibitor, together with the anti-pS294 mAb, inhibits pS294 formation and suppresses ER-dependent gene expression in wild type (ER+) or (ER mutant+) BC. They created ER mutations Ermut (D538G) and (Y537S) in MCF7 cells and illustrated their capacity to trigger tamoxifen-resistant ligand-independent growth, along with the expression of the CDK2-dependent pS294. After the growth of E2-independent and tamoxifen (TAM) resistant MCF7 ERmut (Y537S) tumors in the nude mice, treatment was initiated with different concentrations of dinaciclib or palbociclib. Scott et al concluded that the combined treatment with TAM and palbociclib suppresses tumor growth without any effect on pS294 formation, whereas the TAM/Dinaciclib combination arrests tumor growth and inhibits pS294 expression.141 Dinaciclib is a CDK1 and 2 inhibitor that greatly sensitizes the wild-type BRCA and primary/resistance-mutated BRCA model of TNBC for PARP inhibition. Moreover, dinaciclib increases the response to PARP inhibition in homologous recombination (HR)-deficient cancer.142 Further, dinaciclib treatment leads to an increase in the cleaved caspase 3 and cleaved PARP, downregulates the Mcl1 gene, induces apoptosis, and decreases the number of inflammatory BC cells in a dose-dependent manner. Similarly, dinaciclib enhances the activity of DNA-damaging chemotherapeutic agents, while paclitaxel lacks this ability.143 Another study found that 3-phosphoinositide-dependent protein kinase 1 (PDK1) modulates ribociclib sensitivity in (ER+) MCF-7 cells. Therefore, PDK1 inhibition with GSK2334470 or siRNA in combination with CDK4/6 inhibitor ribociclib or palbociclib suppresses the proliferation and promotes apoptosis in (ER+) BC cells. On the other hand, Ribociclib-resistant cells upregulate the expression of phospho CDK2, cyclins D1, A, E, and phospho-S477/T479 AKT. Therefore, treatment with CDK2 inhibitor dinaciclib or GSK2334470 restores the susceptibility of ribociclib-resistant cells to CDK4/6 inhibition. The combination of ribociclib, GSK2334470, and PI3Kα inhibitor alpelisib decreases tumor growth in the xenograft model. According to research, the PI3K/PDK1 signaling pathway leads to resistance to the CDK4/6 inhibitor through the upregulation of CDKs and S-phase cyclins. Adding dinaciclib to GSK2334470/ribociclib does not increase cell proliferation inhibition.144 Most patients with BC are positive for the hormone receptor and need anti-hormonal treatments such as AI. Reports indicate that low molecular weight (LMW)-E-dependent cell cycle deregulation causes AI resistance, and a combination of CDK2 inhibitors and letrozole can be used to treat patients with (HR+) tumors expressing LMW-E. MCF7 xenografts containing LMW-E are resistant to letrozole, and the tumor volume increases after treatment with AI. Therefore, CDK2 inhibitor dinaciclib reversed LMW-E–dependent resistance, but treatment with CDK4/6 inhibitor Palbociclib does not cause any sensitization.145 Zhu et al reported that the expression of BRCA1 and BARD1 in tamoxifen-resistant mammary tumor cells dramatically increases and leads to resistance to DNA-damaging chemotherapeutic agents such as adriamycin and cisplatin. Hence, the knockdown of the BRCA1 or BARD1 gene or the phosphorylation inhibition of BRCA1 by dinaciclib enhances sensitivity to cisplatin in tamoxifen-resistant cells. Further, the activity of the PI3K/AKT pathway upregulates the expression of BRCA1 and BARD1 genes. Thus, the use of PI3K inhibitors reduces the expression of BRCA1 and BARD1 in tamoxifen-resistant cells and re-sensitizes them to cisplatin.146 It has also been indicated that MAPK signaling plays a key role in regulating cell proliferation, drug resistance, and oncogenic transformation. Trametinib, dabrafenib, and lapatinib participate in the positive regulation of the MAPK pathway, while dinaciclib and rigosertib negatively regulate the MAPK pathway and arrest the cell cycle. Dinaciclib and rigosertib also enhance the phosphorylation of MEK1/2, ERK1/2, and MAPK pathways in BC cells.147 The finding of another study revealed that treatment with AZD1775 results in cleaved PARP and G1 phase accumulation in cyclin E-induced 76NE6 and MDA231 cells. The overexpression of cyclin E causes the stress of DNA replicative and increases DNA double-strand breaks, which can be repaired by activating the Wee1 kinase and ATR-CHK1 pathway. However, after the inhibition of Wee1 kinase by AZD1775 in the cyclin E-high cells, DNA damage repair is not performed enough and cell death occurs due to the accumulation of abnormal nuclei. The combination therapy of AZD1775 and dinaciclib sensitizes cyclin E-low TNBC cells for the inhibition of Wee1 kinase.148 Carey et al showed that MYC-positive tumors were involved in the upregulation of the HR DNA and resistance to DNA-damaging agents such as PARP inhibitors. MYC regulates HR through DNA repair proteins, including RAD51, and inhibition of MYC expression causes susceptibility to PARPi independent of BRCA status. Therefore, the pharmacological inhibition of PARP along with the siRNA knockdown of RAD51 and MYC decreases cell proliferation and increases apoptosis in SUM149 and MB231 cells. The use of dinaciclib and PARP inhibitor niraparib increases DNA damage and decreases the HR and epithelial-mesenchymal transition. Furthermore, Dinaciclib downregulates the expression of the MYC gene and re-sensitizes niraparib-resistant TNBC cells.149 The inhibition of CDK2 and the enhancer of zeste homolog 2 (EZH2) by CDK2i and EZH2i at a lower dose than recommended phase 2 dose (RP2D) induces the expression of ER-α in TNBC cells. Studies have shown that cytokine/chemokine-coding genes such as CCL20, CXCL2, and TNFα, which are upregulated by CDK2 and EZH2i, affect the tumor microenvironment. Likewise, IL-36 cytokine is upregulated by CDK2i/EZH2i and has wide anti-tumor effects. CDK2i/EZH2i also downregulates the RPL39 gene and decreases metastasis and TNBC expansion. CDK2 or EZH2 inhibition leads to re-expressed ER-α and converts TNBC to luminal ER-α-positive, increasing the sensitivity of TNBC cells to tamoxifen. Moreover, the combination therapy with the EZH2 or CDK2 inhibitor and tamoxifen effectively inhibits tumor growth and increases the survival of the mice with TNBC tumors150 (Table 3, Figure 2).
Table 3.
Studies Related to the Effect of Dinaciclib in the Treatment of Breast Cancer
|
Cell Lines
|
Drug/SiRNA
|
Human/Animal Model
|
In vivo/In vitro
|
Study Result
|
Reference
|
| - |
Dinaciclib, capecitabine |
Humans |
In vivo |
Dinaciclib therapy has anti-tumor activity and is commonly safe and tolerable, but its efficacy is less than Capecitabine |
72
|
| - |
Dinaciclib, epirubicin |
Humans |
In vivo |
The combination of dinaciclib and epirubicin has significant toxicity and is not an effective treatment option for TNBC |
140
|
| WHIM3 WHIM12,MDA-MB-231 BT549,HCC1806,BT474, MCF7 T47D |
Dinaciclib, CDK1 siRNA, CDK2 siRNA, CDK9 siRNA, Cyclin B siRNA, |
NOD/SCID mice |
In vitro |
Dinaciclib targets cyclin B1 via the MYC/CDK9 axis and suppresses tumor growth in the TNBC PDX model in vivo |
68
|
| MCF7,SUM44 |
Dinaciclib,palbociclib, SNS-032, JNJ7706621, BMS-265246, tamoxifen, β-estradiol (E2), CDK1 siRNA, CDK2 siRNA, DK9 siRNA |
Nude mice |
In vitro/In vivo |
The combined treatment with tamoxifen and palbociclib could inhibit tumor growth without any effect on pS294 formation, whereas the TAM/dinaciclib combination arrests tumor growth and inhibits pS294 expression |
141
|
| MDA-MB-436,MDA-MB-231,MCF-7,HCC1395,SUM149PT,HCC1937,Hs578T,HCC38,BT549,BT20 |
Dinaciclib, flavopiridol, veliparib, olaparib, Cisplatin
SiBRCA1, SiBRCA2, SiPALB2 |
NOD-SCID-IL2Rγc–/– mice, NMRI-Foxn1nu mice |
In vivo/In vitro |
The combination of the PARP inhibitor and dinaciclib, and DK12 inhibitor could reverse resistance to PARP inhibition in BRCA-mutated cancer |
142
|
| SUM149,HCC1806,HCC1937,MDA-MB-468,BT549, MCF-7,MDA-MB-157,MDA-MB-231,MDA-MB-436,SUM185, KPL4,SKBR3, T47D,MDA-MB-361 |
Dinaciclib, epirubicin, carboplatin, paclitaxel |
Humans |
In vivo/In vitro |
Treatment with Dinaciclib augments the cleaved PARP/caspase 3, downregulates the Mcl1 expression, and promotes apoptosis |
143
|
| MCF-7, T47D HCC1428,HCC1500 |
Dinaciclib, abemaciclib, palbociclib, ribociclib, PI3Kα inhibitor: alpelisib, GSK2334470, siPDK1 |
Ovariectomized athymic mice |
In vivo/In vitro |
Treatment with GSK2334470 or CDK2 inhibitor Dinaciclib restores the susceptibility of Ribociclib-resistant cells to CDK4/6 inhibition |
144
|
| Aromatase-overexpressing LMW-E MCF7/T47D |
Aromatase inhibitors: letrozole, exemestane, anastrozole, doxycycline |
Nude mice
Humans |
In vivo/In vitro |
Dinaciclib reversed LMW-E–dependent AI resistance |
145
|
| MCF7, T47D |
Dinaciclib, cisplatin, paclitaxel, adriamycin, PI3K inhibitor: BKM120, BRCA1 siRNA, BARD1 siRNA |
NOD/SCID mice |
In vivo/In vitro |
Dinaciclib inhibits the phosphorylation of BRCA1 and re-sensitizes Tamoxifen-resistant cells to cisplatin |
146
|
| MCF-10A, MCF-7MCF12A, BT474,MDA-MB-231,MDA-MB-453,MDA-MB-468 |
Dinaciclib, rigosertib (polo-like kinase 1 inhibitor), dabrafenib, trametinib |
- |
In vitro |
Dinaciclib and Rigosertib negatively regulate the MAPK pathway and arrest the cell cycle |
147
|
| 76NE6 cyclin E–inducible,HEK-293T,HCC1806,MDA436,MDA468 MDA231, SUM149, SUM159 |
DinaciclibAZD1775 (Wee1 kinase inhibitor), carboplatin |
PDX models (mice) |
In vivo/In vitro |
Combination therapy of AZD1775 and Dinaciclib sensitizes cyclin E-low TNBC cells to the inhibition of Wee1 kinase |
148
|
| MDA-MB-231,MDA-MB-468, HCC1937, HCC1806,SUM149,SUM1315,MDA-MB-436,MDA-MB-157,MCF-10A,PC3, DU145 A549, Calu-1,H1299, H1993,OVCAR3, 59M FUOV1, BxPC3, PANC-1, and HCT116 |
Dinaciclib,
Niraparib, MYC siRNA, RAD51 siRNA |
Nude mice |
In vivo/In vitro |
Dinaciclib and PARP inhibitor niraparib increase DNA damage while decreasing the homologous recombination and epithelial-mesenchymal transition. Dinaciclib downregulates the expression of the MYC gene and re-sensitizes niraparib-resistant TNBC cells |
149
|
MDA-MB-231 SUM-149
BT549, T47D |
Dinaciclib, EPZ-6438 Tamoxifen |
NOD/SCID mice |
In vivo/In vitro |
A combination of CDK2i Dinaciclib and EZH2 leads to re-expressed ER-α, converts TNBC to luminal ER-α-positive, and increases the sensitivity of TNBC cells to Tamoxifen |
150
|
Note. Small interfering RNA; TNBC: Triple-negative breast cancer; PARP: Poly (ADP-ribose) polymerase; BRCA: Breast cancer gene; LMW: Low molecular weight; AI: Aromatase inhibitor; ER: Estrogen receptor; PDX: Patient-derived xenograft; MAPK: Mitogen-activated protein kinase.
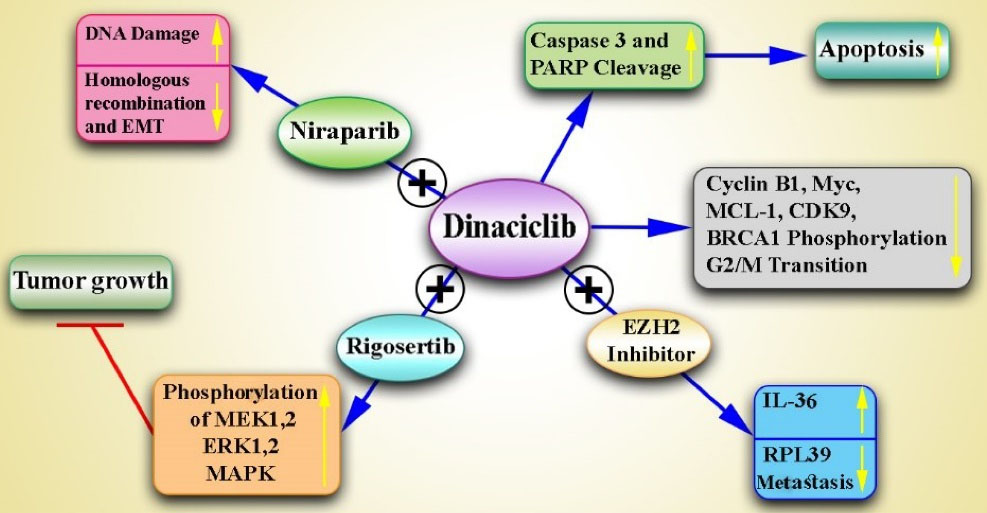
Figure 2.
Combination of Dinaciclib With Other Drugs for Breast Cancer Therapy. Note. Dinaciclib reduces cyclin B1 and Myc expression through CDK9 inhibition, leads to the arrest of the G2/M phase cell cycle, increases cleaved caspase 3 and cleaved PARP, downregulates the Mcl1 gene, inhibits the phosphorylation of BRCA1, and induces apoptosis. The combination of Dinaciclib and Rigosertib also enhances the phosphorylation of the MEK1/2, ERK1/2, and MAPK pathway in breast cancer cells. Dinaciclib and Niraparib increase DNA damage while decreasing the homologous recombination and epithelial-mesenchymal transition. Dinaciclib and EZH2i upregulate IL-36 cytokine, downregulate the RPL39 gene, and decrease metastasis. PARP: Poly (ADP-ribose) polymerase; MCL-1: Myeloid cell leukemia 1; MEK: Mitogen-activated protein kinase, ERK: Extracellular-signal-regulated kinase; MAPK: Mitogen-activated protein kinase; EZH2i: Enhancer of zeste homolog 2 inhibitor.
.
Combination of Dinaciclib With Other Drugs for Breast Cancer Therapy. Note. Dinaciclib reduces cyclin B1 and Myc expression through CDK9 inhibition, leads to the arrest of the G2/M phase cell cycle, increases cleaved caspase 3 and cleaved PARP, downregulates the Mcl1 gene, inhibits the phosphorylation of BRCA1, and induces apoptosis. The combination of Dinaciclib and Rigosertib also enhances the phosphorylation of the MEK1/2, ERK1/2, and MAPK pathway in breast cancer cells. Dinaciclib and Niraparib increase DNA damage while decreasing the homologous recombination and epithelial-mesenchymal transition. Dinaciclib and EZH2i upregulate IL-36 cytokine, downregulate the RPL39 gene, and decrease metastasis. PARP: Poly (ADP-ribose) polymerase; MCL-1: Myeloid cell leukemia 1; MEK: Mitogen-activated protein kinase, ERK: Extracellular-signal-regulated kinase; MAPK: Mitogen-activated protein kinase; EZH2i: Enhancer of zeste homolog 2 inhibitor.
SNS-032
E2F1 and E2F4 play an essential role in cell cycle regulation. Ma et al investigated the role of E2F1/E2F4 in apoptosis resulting from CDK inhibitors (BMS-387032, roscovitine, and flavopiridol) and chemotherapeutic agents (paclitaxel, cisplatin, and VP16). They found that treatment with these CDK inhibitors and chemotherapeutic drugs reduces the level of E2F4. Furthermore, reducing E2F4 through RNAi increases the susceptibility of human cancer cells to the drug.151 In another study, they indicated that treatment with BMS-387032 stabilizes E2F1 protein by the increase in the expression of p57 protein/mRNA, but suppresses the E2F1 mRNA and some E2F family members in MDA-MB-231 BC cells.152 Phosphorylated ER-α is considered a biomarker and a predictive/therapeutic value for (ER-α +) BCs. Held et al showed that the Ser294 phosphorylation of ER-α in human BC cells is conducted by the ligand (tamoxifen and estradiol) stimulation of ER-α rather than the growth factor activation (heregulin-β, EGF, and insulin) of ER-α. The use of SNS-032 or roscovitine prevents the Ser294 phosphorylation of ligand-activated without affecting the phosphorylation of other serins (Ser104/106 or Ser118).153 It has also been represented that SNS-032 induces apoptosis through both internal and external apoptotic pathways and increases the activation of poly (ADP-ribose) polymerase (PARP) and caspase 8/9 in BC cells. It also promotes RNA polymerase II dephosphorylation in serines 2 and 5 and inhibits RNA synthesis, reduces Mcl-1 and XIAP in BC cells, and represses the mammary tumor growth in mice.154 ATP-binding cassette transporters cause multidrug resistance (MDR) toward chemotherapeutic/anticancer drugs. Cihalova et al assessed the interaction of CDK inhibitors (SNS-032, AT-7519, and flavopiridol) with ABC transporters (ABCB1, ABCG2, and ABCC1) under laboratory conditions. They reported that SNS-032 decreases ABCG2-dependent efflux. In addition, flavopiridol could inhibit ABCC1 and ABCG2, whereas AT-7519 had no effect on the inhibition of ABCG2, ABCC1, or ABCB1. Additionally, SNS-032 and flavopiridol in combination with the substrates of the ABC transporter such as topotecan and daunorubicin had anti-proliferative effects on cancer cells.155 It was also revealed that PCTK1, PCTAIRE1, and Cdk16 play a fundamental role in tumor formation and cancer cell resistance to TNF-family cytokines, and their expression in cancer cells represents an increase. The protein kinase PCTAIRE1 takes part in the extrinsic pathway of apoptosis. The knockdown of the PCTAIRE1 gene using shRNA induces the degradation of RIPK1 and caspase-8 cleavage without any effect on the cell cycle arrest and sensitizes breast and prostate cancer cells to TNF-family cytokines, including TRAIL. Further, SNS-032 suppresses the function of PCTAIRE1 kinase and sensitizes PPC1 prostate cancer cells to TRAIL-mediated apoptosis156 (Table 4, Figure 3).
Table 4.
Studies Related to the Role of SNS-032 and Purvalanol A in Breast Cancer
|
Cell Lines
|
Drug/SiRNA
|
Human/Animal Model
|
In vivo/In vitro
|
Study Result
|
Reference
|
| H1299, MDA-MB-231 MCF7, 3T3 |
Flavopiridol, roscovitine, BMS-387032, cisplatin, VP16, paclitaxel, E2F1 RNAi |
- |
In vitro |
Treatment with CDK inhibitors and chemotherapeutic drugs decreases the level of E2F4 |
151
|
H1299, MCF-7 MDA-MB-231 T98G
(wt Rb, p53, and mut p16) |
BMS-387032, p57RNAi |
- |
In vitro |
Treatment with BMS-387032 stabilizes the E2F1 protein but suppresses E2F1 mRNA in the MDA-MB-231 breast cancer cells |
152
|
| MCF-7, BT474, T47D |
SNS-032, β-estradiol, tamoxifen, okadaic acid, forskolin, insulin, heregulin-β, IGEPAL CA-630 |
- |
In vitro |
The use of SNS-032 or roscovitine prevents from the Ser294 phosphorylation of ligand-activated in ER-α |
153
|
| MCF-7, MDA-MB-435 |
SNS-032 |
BALB/c-nu mice |
In vivo/In vitro |
SNS-032 induces apoptosis through both internal and external apoptotic pathways in breast cancer cells |
154
|
MDCKII (MDCKII-ABCB1, MDCKII-ABCG2, and MDCKII-ABCC1),
HepG2, T47D |
SNS-032 AT-7519, Flavopiridol, Daunorubicin, Mitoxantrone, MK-571 (ABCC1 inhibitor), LY335979 (ABCB1 inhibitor), Ko143, (ABCG2 inhibitor) |
- |
In vitro |
SNS-032 decreases ABCG2-dependent efflux
SNS-032 and flavopiridol in combination with the substrates of the ABC transporter such as topotecan and daunorubicin have antiproliferative effects on cancer cells |
155
|
| PPC1, Du145, MDA-MB-468, T47D, MCF7 IMR-90, HeLa HEK293T, 267B1, 267B1/K-ras |
SNS-032, SiPCTAIRE1 SiPCTAIRE2 SiPCTAIRE3, Sicaspase-8 SiRIPK1 |
- |
In vitro |
SNS-032 suppresses the acting of PCTAIRE1 kinase and sensitizes PPC1 prostate cancer cells to TRAIL-mediated apoptosis |
156
|
| MCF7, MELN (MCF-7 ERE-beta-globin Luciferase Neo), MCF-7/mdr1, HT-29, A31, BP-A31 |
Purvalanol A |
- |
In vitro |
Porvalvanol A inhibits Rb phosphorylation in the G1 stage, while its inhibitory activity is weak toward cdk4/cyclin D1 |
157
|
| MDA MB 231, HeLa, A2780, SKOV3, DRO90-1, ARO81-1, HCT116, HCT116 p21-/- |
Purvalanol A, roscovitine, paclitaxel, cyclin B siRNA, Histone deacetylase inhibitor |
- |
In vitro |
Combination therapy with Purvalanol A, Roscovitine, histone deacetylase inhibitor, and downregulation of cyclin B using siRNA decreases the hyperphosphorylation of 4E-BP1 in cell treated with PTX |
158
|
| MDCKII (MDCKII-ABCG2) |
Purvalanol A, lomoucine II, roscovitine, bohemine,olomoucine, mitoxantrone, fumitremorgin C (ABCG2 inhibitor) |
Wistar rats |
In vivo/In vitro |
A combination of Purvalanol A and Olomoucine II can strengthen the cytostatic effect of mitoxantrone (ABCG2 substrate) in cells expressing ABCG2 |
159
|
| MCF-7, MDA-MB 231 |
Purvalanol A, MDL72,527 (PAO/SMO inhibitor) |
- |
In vitro |
Purvalanol A causes mitochondria-induced apoptosis and modulates anti- and pro-apoptotic proteins and the polyamine catabolic pathway in MDA-MB-231 and MCF-7 cells |
160
|
Note. PAO: Polyamine oxidase; SMO: Spermine oxidase; CDK: Cyclin-dependent kinase; SiRNA: Small interfering RNA; ER: Estrogen receptor; PTX: Paclitaxel; TRAIL: Tumor necrosis factor-related apoptosis-inducing ligand.
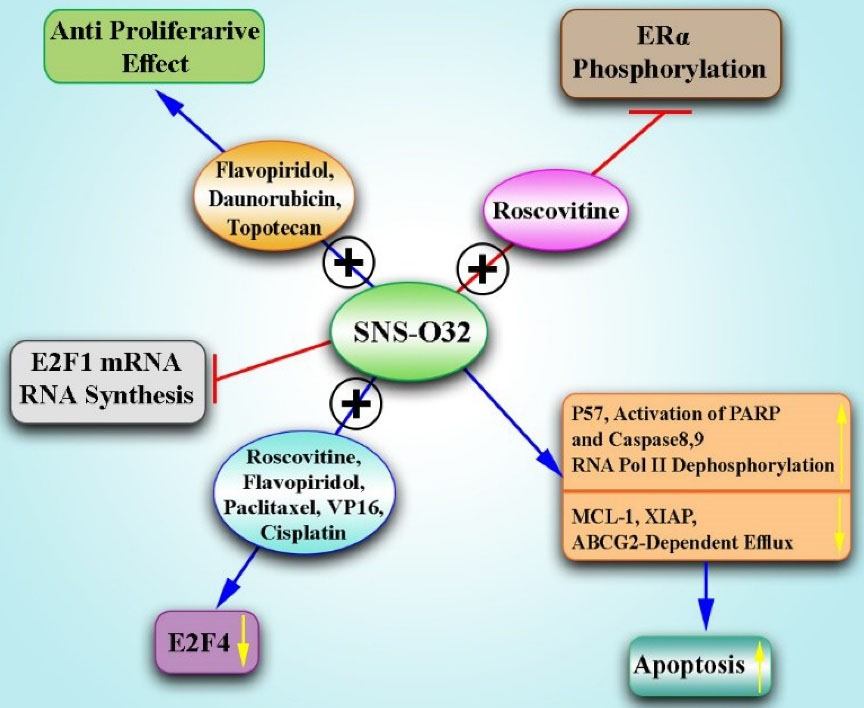
Figure 3.
Combination of SNS-032 With Other Therapeutics for BC Therapy.Note. Treatment with BMS-387032 stabilizes E2F1 protein by the increase in the expression of p57 protein/mRNA but suppresses E2F1 mRNA in BC cells. In addition, SNS-032 induces apoptosis through both internal and external apoptotic pathways, and increases the activation of PARP, and caspase 8/9 in BC cells. It also promotes RNA polymerase II dephosphorylation in serines 2 and 5 and inhibits RNA synthesis, reducing Mcl-1, XIAP, and ABCG2-dependent efflux in BC cells. Moreover, the use of SNS-032 or roscovitine prevents the Ser294 phosphorylation of ER-α. A combination of BMS-387032, Roscovitine, and Flavopiridol with chemotherapeutic agents (Paclitaxel, Cisplatin, and VP16) decreases the level of E2F4. In addition, SNS-032 and Flavopiridol in combination with Topotecan and Daunorubicin have antiproliferative effects on cancer cells. ATP: Anion transporter polypeptide; PARP: Poly (ADP-ribose) polymerase; MCL-1: Myeloid cell leukemia 1, XIAP: X-linked inhibitor of apoptosis protein; ABCG2: ATP-binding cassette super-family G member 2; BC: Breast cancer.
.
Combination of SNS-032 With Other Therapeutics for BC Therapy.Note. Treatment with BMS-387032 stabilizes E2F1 protein by the increase in the expression of p57 protein/mRNA but suppresses E2F1 mRNA in BC cells. In addition, SNS-032 induces apoptosis through both internal and external apoptotic pathways, and increases the activation of PARP, and caspase 8/9 in BC cells. It also promotes RNA polymerase II dephosphorylation in serines 2 and 5 and inhibits RNA synthesis, reducing Mcl-1, XIAP, and ABCG2-dependent efflux in BC cells. Moreover, the use of SNS-032 or roscovitine prevents the Ser294 phosphorylation of ER-α. A combination of BMS-387032, Roscovitine, and Flavopiridol with chemotherapeutic agents (Paclitaxel, Cisplatin, and VP16) decreases the level of E2F4. In addition, SNS-032 and Flavopiridol in combination with Topotecan and Daunorubicin have antiproliferative effects on cancer cells. ATP: Anion transporter polypeptide; PARP: Poly (ADP-ribose) polymerase; MCL-1: Myeloid cell leukemia 1, XIAP: X-linked inhibitor of apoptosis protein; ABCG2: ATP-binding cassette super-family G member 2; BC: Breast cancer.
Purvalanol A
Villerbu et al found that purvalanol A causes cell cycle arrest and selective phosphorylation inhibition of several CDK substrates in synchronized cells. Prolonged contact of growing cells with this drug causes the stable inhibition of proliferation and cell death. Furthermore, purvalanol A inhibits Rb phosphorylation in the G1 stage, while its inhibitory activity is weak toward cdk4/cyclin D1. Additionally, the drug is unable to inhibit the activity of cdk7, an essential kinase for the activation of Thr172 phosphorylation in cdk4.157 In another study, it was demonstrated that paclitaxel induces the hyperphosphorylation of the 4E-binding protein (4E-BP1) and decreases its association with eIF-4E in the MDA-MB231 cancer cells, but it does not affect phosphorylation or expression of eukaryotic initiation factor 4E (eIF-4E). It is noteworthy that 4E-BP1 phosphorylation is associated with the accumulation of G2/M and increased phosphorylation of cdk1 substrates. Combination therapy with purvalanol A, roscovitine, histone deacetylase inhibitor, which indirectly inhibits CDK function, and downregulation of cyclin B using siRNA reduces the hyperphosphorylation of 4E-BP1 in cells treated with paclitaxel. However, monotherapy with paclitaxel escalates the eIF-4E level by raising the hyperphosphorylation of 4E-BP1 via a cdk1-dependent process.158 A combination of purvalanol A and olomoucine II can strengthen the cytostatic effect of mitoxantrone (ABCG2 substrate) in cells expressing ABCG2. In addition, the concomitant use of purvalanol A or olomoucine II with other anti-cancer drugs, that target ABCG2, can lead to cytostatic effects, which is an effective cancer therapy for predominating MDR.159 Purvalanol A also promotes apoptosis in a caspase-dependent manner in (ER + ) MCF-7 cells, whereas (ER-) MDA-MB-231 cells are less sensitive to the drug. Further, cells that highly express Bcl-2 are more resistant to purvalanol A-induced apoptosis. Moreover, Purvalanol A triggers the upregulation of spermidine/spermine N1-acetyltransferase (SSAT) and polyamine oxidase (PAO) in MCF-7 cells. The use of PAO/spermine oxidase inhibitor (MDL 72,527) impedes purvalanol A-mediated apoptosis. Generally, purvalanol A causes mitochondria-induced apoptosis, reduces cell viability, and modulates anti- and pro-apoptotic proteins and the polyamine catabolic pathway in MDA-MB-231 and MCF-7 cells160 (Table 4, Figure 4).
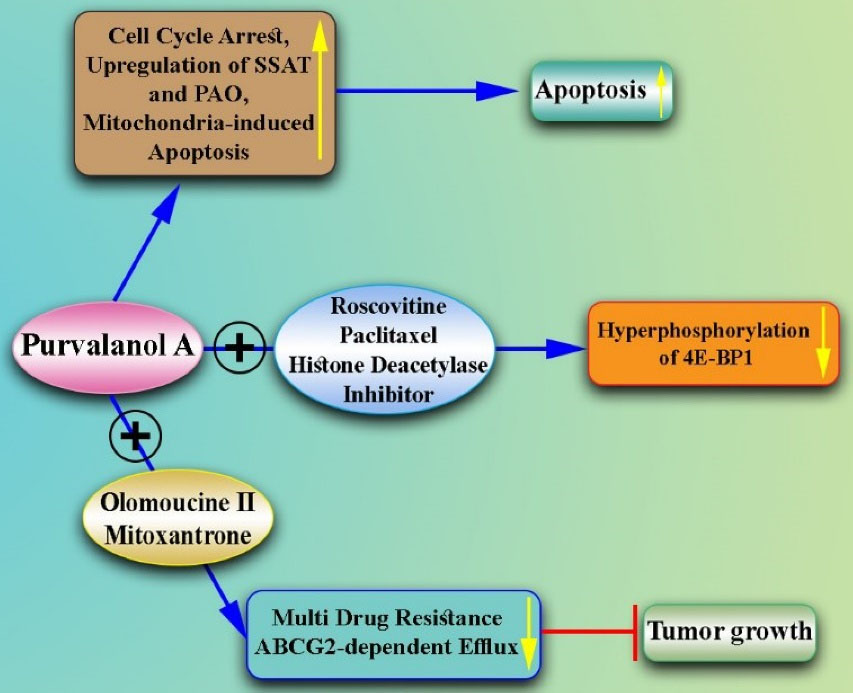
Figure 4.
Combination of Purvalanol A With Other Drugs for the Treatment of Breast Cancer. Note. Purvalanol A causes cell cycle arrest, inhibits Rb phosphorylation in the G1 stage, and promotes apoptosis. Further, it triggers the upregulation of SSAT and PAO in MCF-7 cells. Furthermore, Purvalanol A causes mitochondria-induced apoptosis and modulates anti- and pro-apoptotic proteins and the polyamine catabolic pathway in breast cancer cells. The combination therapy of Purvalanol A, Roscovitine, and histone deacetylase inhibitor decreases the hyperphosphorylation of 4E-BP1 in cells treated with Paclitaxel. Moreover, the combination of Purvalanol A and Olomoucine II can strengthen the cytostatic effect of mitoxantrone (ABCG2 substrate) in cells expressing ABCG2, which is an effective cancer therapy for predominating multidrug resistance. SSAT: Spermidine/spermine N1-acetyltransferase; PAO: Polyamine oxidase; 4E-BP1: 4E-binding protein; ABCG2:ATP-binding cassette super-family G member 2.
.
Combination of Purvalanol A With Other Drugs for the Treatment of Breast Cancer. Note. Purvalanol A causes cell cycle arrest, inhibits Rb phosphorylation in the G1 stage, and promotes apoptosis. Further, it triggers the upregulation of SSAT and PAO in MCF-7 cells. Furthermore, Purvalanol A causes mitochondria-induced apoptosis and modulates anti- and pro-apoptotic proteins and the polyamine catabolic pathway in breast cancer cells. The combination therapy of Purvalanol A, Roscovitine, and histone deacetylase inhibitor decreases the hyperphosphorylation of 4E-BP1 in cells treated with Paclitaxel. Moreover, the combination of Purvalanol A and Olomoucine II can strengthen the cytostatic effect of mitoxantrone (ABCG2 substrate) in cells expressing ABCG2, which is an effective cancer therapy for predominating multidrug resistance. SSAT: Spermidine/spermine N1-acetyltransferase; PAO: Polyamine oxidase; 4E-BP1: 4E-binding protein; ABCG2:ATP-binding cassette super-family G member 2.
Roscovitine
As mentioned in the previous section, roscovitine can be considered a potent anti-cancer therapeutic when administrated alone or in combination with other anti-tumor therapeutics. Several studies have confirmed the effectiveness of this CDK inhibitor in the treatment of BC, which will be discussed in this section. Mgbonyebi et al first reported the pro-apoptotic and anti-proliferative impact of roscovitine on MDA-MB-231 and MCF-7 BC cells in vitro.161,162 Wojciechowski et al attempted to clarify the pro-apoptotic and anti-proliferative effects of roscovitine in MCF-7 cells. They showed that treatment of cancer cells with roscovitine modifies the pattern of the argyrophilic nucleolar organizer regions and nucleolar RNA in cancer cells in a time-dependent manner. This was associated with the increased number of nucleolar fragments, redistribution of the nucleolin, and reduced histone phosphorylation and acetylation. These changes arrested cancer cells at the G2/M phase, increased p53 levels, and induced apoptosis, which was related to the upregulation of caspases. Therefore, it was proposed that roscovitine promotes the apoptosis of cancer cells through the primary disintegration of nucleoli and cell cycle arrest.163 Roscovitine upregulates p53 partly through the repression of its negative regulator (MDM2). It was hypothesized that the degradation of p53 by proteasome inhibitors may mimic the effects of roscovitine. However, the examination of this hypothesis in MCF-7 cells revealed that while roscovitine arrests the cell cycle at the G2 phase, proteasomes do it at the S phase. Thus, although proteasome inhibitors could increase p53 expression, they did not mimic roscovitine effects.164 Wesierska-Gadek et al also demonstrated the mechanism by which roscovitine upregulates p53 in cancer cells. They concluded that the treatment of MCF-7 cells with roscovitine leads to the activation and overexpression of homeodomain-interacting protein kinase-2 (HIPK2). Subsequently, HIPK2 promotes the basal and roscovitine-mediated phosphorylation of p53 at Ser-46, thereby stabilizing wild-type p53 and enhancing roscovitine-mediated apoptosis in cancer cells.165
Interestingly, it has been represented that the effect of roscovitine on the proliferation and apoptosis of MCF-7 cancer cells depends on the cell cycle status in vitro. This claim was proved by the exposure of G1 phase-arrested (through serum withdrawal) and S phase-arrested (by hydroxyurea) cells to roscovitine. The treatment of G1-synchronized cancer cells with roscovitine led to increased cell cycle arrest and decreased number of S-phase cells. A lesser outcome was obtained following the treatment of MCF-7 cells rescued from G1 phase arrest for 4 hours with roscovitine. On the other hand, the treatment of cells rescued from G1 arrest for 24 hours with roscovitine increased the frequency of G1 cells. Roscovitine could also increase S phase cells in MCF-7 cells rescued from S phase arrest. Therefore, based on the cell cycle status, roscovitine differentially affects the proliferation of cancer cells.166 Similarly, it was found that the outcome of the treatment of BC cells (MCF-7) with roscovitine critically relies on the cell cycle status and drug dose. Although roscovitine mainly suppresses CDKs at low doses, which leads to cell cycle arrest, it induces apoptosis at high doses.167
In another attempt, the anti-proliferative effect of roscovitine was evaluated in MCF-7 cells in vitro. The treatment of cancer cells with roscovitine led to cell cycle arrest at the G2/M phase after 6 hours, which was associated with the upregulation of p53. Roscovitine induced apoptosis in MCF-7 cells through the mitochondrial pathway.99 Furthermore, the blockade of farnesyl protein transferase enzyme in MCF-7 cells increased the anti-proliferative impact of roscovitine in these cells in vitro. The overexpressed c-Ha-Ras protein in MCF-7 cells needs to be farnesylated by the farnesyl protein transferase to achieve full biological function. The exposure of MCF-7 cells to roscovitine in combination with L-744,832, an inhibitor of farnesyl protein transferase, markedly induced higher apoptosis in cancer cells compared to monotherapy with either roscovitine or L-744,832. Similarly, combined therapy potently reduced cancer cell proliferation in comparison to monotherapy.168 Downregulation of phosphatase nuclear targeting subunit (PNUTS) also enhanced roscovitine-mediated apoptosis in MCF-7 cells. The phosphorylation status of Rb plays an important role in the modulation of apoptosis and proliferation in cancer cells. CDKs promote Rb phosphorylation following growth signals, while protein phosphatase 1 (PP1) enhances Rb dephosphorylation through E2F1 following pro-apoptotic and anti-proliferative signals. The knockdown of PNUTS (PP1 interacting protein) promotes the dephosphorylation of Rb and apoptosis in MCF-7 cells. Accordingly, the dephosphorylation of Rb by roscovitine or siRNA mediated the knockdown of PNUTS led to increased apoptosis in cancer cells up to two folds. Combined treatment by roscovitine and PNTUS-specific siRNA further enhanced apoptosis up to four folds.169
It has been reported that the anti-proliferative and pro-apoptotic effects of roscovitine are mainly through targeting CDK5 activity in highly invasive MDA-MB231 cells in vitro. Goodyear et al demonstrated that bFGF increases CDK5 expression and CDK5 regulates the proliferation of MDA-MB231 cells. They indicated that roscovitine selectively suppresses CDK5, resulting in the induction of apoptosis and prevention of cancer cell proliferation.170 Regarding the importance of CDK5 activity in tumor-initiating cells, it is also assumed that the blockade of this CDK can suppress the development of these cells. Accordingly, the blockade of CDK5 by roscovitine or siRNA technology suppressed sphere formation and tumor establishment in vivo. Mandl et al showed that the blockade of CDK5 suppressed tumorsphere formation through inducing the Foxo1 transcription factor and thereby upregulating Bim pro-apoptotic protein.171
It is proposed that roscovitine can promote the apoptosis process in MCF-7 cells through different pathways, relying on the caspase 3 status in these cells. Roscovitine promoted the mitochondrial pathway of apoptosis in caspase 3-deficient MCF-7 cells through p53 phosphorylation at Ser46, leading to the upregulation of p53AIP1 protein and thus the depolarization of mitochondrial potential. On the other hand, roscovitine could not promote p53 Ser46 phosphorylation in caspase-3-expressing cells.98 However, the forced expression of caspase 3 in MCF-7 cells did not affect their sensitization to roscovitine-induced apoptosis.172 It is also reported that the blockade of ornithine decarboxylase, which is overexpressed in BC cells, can affect roscovitine-mediated apoptosis through the mitochondrial pathway in MCF-7 cells. Ornithine decarboxylase regulates the concentration of polyamines in cells. It has been demonstrated that the blockade of ornithine decarboxylase by its inhibitor (α-difluoromethylornithine) suppresses the proliferation of BC cells. The exposure of MCF-7 cells to α-difluoromethylornithine changed the pro-apoptotic effects of roscovitine through reducing polyamine levels, inducing reactive oxygen species, and regulating the expression of Bcl-2 family proteins.173 Interestingly, exogenous polyamines play a pivotal role in the induction of apoptosis or autophagy by roscovitine in MCF-7 (ER+) and MDA-MB-231 (ER-) cells. MDA-MB-231 cells represented higher resistance to roscovitine compared to MCF-7 cells, which was partly due to the regulation of the autophagy pathway in ER- cells.174
In addition to pro-apoptotic action, it is found that roscovitine can sensitize cancer cells against radiation both in vitro and in vivo. The treatment of MDA-MB 231 cells with roscovitine in combination with ionizing radiation led to a robust increase in micronuclei frequency and impairment in DNA-double-strand break repair compared to cells treated with ionizing radiation alone. A similar defect in the nonhomologous end-joining pathways was detected in BALB murine cells compared to DNA-PKcs-deficient SCID murine cells. Roscovitine-induced radiosensitization was also associated with the blockade of the DNA-dependent protein kinase function triggered via a reduced Ku-DNA binding affinity. Therefore, roscovitine enhances radiosensitization in BC cells lacking a functional p53 partly through an effect on the DNA repair system.175 Interestingly, this combination therapy could also inhibit the angiogenesis process both in vitro and in vivo. This combination approach could significantly reduce microvascular density and angiogenic hot spots in MDA-MB 231 in comparison with either roscovitine or ionizing radiation. Reduced angiogenesis was associated with increased Ang-2/Tie-2, reduced vascular endothelial growth factor (VEGF) levels, and thereby vessel destabilization. Ionizing radiation also enhanced roscovitine-mediated anti-proliferation without triggering apoptosis in endothelial cells. On the other hand, roscovitine suppressed the radiation-induced secretion of VEGF by endothelial and MDA-MB 231 cells. Furthermore, the combination of roscovitine and ionizing radiation robustly inhibited the invasion of endothelial cells and the capillary-like tube formation. The lack of toxicity against normal cells further substantiated the efficacy of this combinatorial treatment.176
Naturally occurring compounds can also enhance the anti-cancer potential of roscovitine.177 Considering that polyphenols such as resveratrol and epicatechins, which are rich in food, exert pro-apoptotic and anti-proliferative effects on cancer cells as anti-oxidant agents, it is assumed that they may improve the anti-cancer effects of roscovitine. Therefore, MCF-7 cells were treated with the combination of roscovitine and resveratrol which was associated with higher pro-apoptotic and anti-proliferative effects compared to cells treated with roscovitine or resveratrol, implying their synergistic effect.178 In addition to the impact of other anti-cancer therapeutics on the efficacy of roscovitine, it can also enhance the anti-cancer activity of some drugs. For instance, it is demonstrated that roscovitine facilitates the apoptosis of BT-474, MDA-MB435-S, SKBr-3, and MDA-MB468 cancer cells by the tumor necrosis factor (TNF)-related apoptosis-inducing ligand (TRAIL). It is reported that BC cells are resistant to TRAIL-mediated apoptosis. Interestingly, the addition of roscovitine to the culture of TRAIL-treated cancer cells resulted in TRAIL-mediated apoptosis in TRAIL-resistant cells. Roscovitine-induced TRAIL sensitivity, partly through the downregulation of FLICE-inhibitory proteins, cFLIP(S), and cFLIP(L), enhances the formation of TRAIL death-inducing signaling complex, activates caspase-8, and downregulates Mcl-1 and E2F1.179 The modulation of Mcl-1 expression by roscovitine also sensitizes BT474 BC cells to BAX/BAK-dependent mitochondrial apoptosis both in vitro and in vivo.180
Moreover, it has been shown that roscovitine can enhance the anti-cancer activity of doxorubicin in MCF-7 cells and MCF7 xenograft-bearing nude mice mainly through increasing cell cycle arrest and to a lesser extent, by inducing apoptosis. Appleyard et al found that combined therapy by roscovitine and doxorubicin significantly reduced the proliferation of cancer cells as assayed by MIB1 (ki67) immunohistochemistry and increased the expression of p21 and p27. However, it did not affect the expression of p53 and survivin and p53 phosphorylation (Ser15).100 This combination strategy could also kill LMV cyclin E-expressing MCF-7 cells in vitro. LMW-E is only expressed in cancer cells, binds to CDK2, and promotes BC progression.181,182 The expression of LMW-E in BC cells triggers resistance to letrozole. Treatment with roscovitine prevented resistance to letrozole in LMW-E-expressing MCF-7 cells.183 Additionally, Jabbour-Leung et al concluded that sequential combined therapy by roscovitine and Doxorubicin is potently lethal in TNBC with p53 mutation. The p53 mutations in TNBC help in the G1 bypass and promote cell cycle progress even in the lack of DNA repair. Thus, it was expected that TNBCs to be sensitive to cell cycle arrest by roscovitine in combination with chemotherapy. Accordingly, the sequential treatment of TNBC cells with doxorubicin after roscovitine resulted in significant apoptosis of cancer cells. Roscovitine arrests the cell cycle in the G2/M phase, leading to DNA damage in TNBC cells. Combined therapy increased DNA damage, which was associated with reduced function of the DNA repair system. Moreover, the sequential administration of roscovitine before doxorubicin into MDA-MB-468 breast tumor-bearing nude mice led to reduced tumor size and increased survival rate, representing that the disruption of p53 signaling is needed to induce cytotoxicity by combination therapy in TNBC, making p53 a prognosticator factor of response to combination treatment.184 Given the relationship between HER2 and cyclin E, it was anticipated that the combination of anti-HER-2 and roscovitine might be an effective strategy for BC therapy.185 Accordingly, a synergistic effect was observed following the treatment of SKBR3 and MCF-7 BC cells with the combination of roscovitine and the anti-HER-2 antibody both in vitro and in vivo.186
Evidence also indicates the role of roscovitine in the induction of apoptosis in drug-resistant P-glycoprotein (P-gp)-overexpressing MCF-7 BC cells both in vitro and in vivo. Drug resistance is usually associated with the upregulation of the MDR1 gene, encoding the P-gp transporter. Caspase 3, which is defective in MCF-7 BC cells, can cleave P-gp. The treatment of P-gp-overexpressing MCF-7 cells with recombinant caspases, 3, 6, and 7 led to the cleavage of P-gp in vitro. On the other hand, the apoptosis of cancer cells by roscovitine was not associated with P-gp cleavage even in the presence of active caspases 6 and 7 in vivo. Surprisingly, P-gp overexpressing cancer cells showed higher sensitivity to roscovitine compared to wild-type cells, highlighting the impact of this CDK inhibitor on P-gp mediated drug resistance.187
It is demonstrated that the apoptotic effects of roscovitine in MCF-7 cells can be potently reduced in the presence of estrogen analogs. Therefore, it is assumed that roscovitine might affect the phosphorylation status of ER-α in these cells. The treatment of MCF-7 cells with roscovitine resulted in the blockade of CDK2 and 7, inhibited RNA polymerase II phosphorylation by CDK7, and decreased basal ER-α phosphorylation (Ser118) in non-stimulated MCF-7 cells. The stimulation of MCF-7 cells with estrogen potently stimulated ER-α by phosphorylation at Ser118, which could be prevented by roscovitine. Tamoxifen (anti-estrogen) could further enhance roscovitine-induced cell cycle arrest at G1 or G2 phases. Hence, roscovitine can inhibit the activating phosphorylation of ER-α and exhibit synergistic effects in combination with anti-estrogen therapy.89 Consistently, the treatment of various ER-α positive or negative BC cells with the combination of roscovitine and tamoxifen synergistically suppressed their proliferation and induced apoptosis in vitro.188-190 In addition, roscovitine can suppress the growth and development of hormonal therapy-resistant BC cells through the blockade of CDK2 both in vitro and in vivo. Nair et al concluded that the activation of the CDK2 pathway is aberrant in three hormonal therapy-resistant cells, including the MCF-7-HER2, MCF-7-LTLTca, and MCF-7-TamR. The treatment of these cells with roscovitine led to cell cycle arrest at the G2 phase, reduced CDK2 activity, downregulated cyclin D1, and reduced the expression levels of ER-α and its coregulators such as PELP1 and AIB1. The administration of roscovitine into xenograft tumor models further substantiated the efficacy of this CDK inhibitor in the suppression of hormone therapy-resistant tumors.114
The PR/cyclin D1 pathway promotes BC progression through cell cycle-dependent transcriptional signals, thus it is assumed that roscovitine can modulate this program. Accordingly, the inhibition of CDK2 by roscovitine in T47D cells blocked the interaction of PR/cyclin D1 with DNA and reduced the expression of HSPB8. As a heat-shock protein, HSPB8 is one of the PR target genes whose upregulation leads to tamoxifen resistance191 (Table 5, Figures 5 and 6).
Table 5.
Studies Related to the Role of Roscovitine in Breast Cancer
|
Cell Lines
|
Drug/SiRNA
|
Animal Model
|
In vivo/In vitro
|
Study Result
|
Reference
|
| MDA-MB-231 |
Roscovitine |
- |
In vitro |
Roscovitine enhances apoptosis in the ER - MDAMB-231 BC cells |
161,162
|
| MCF-7 |
Roscovitine |
- |
In vitro |
Roscovitine induces apoptosis in cancer cells through the primary disintegration of nucleoli and cell cycle arrest |
163
|
| MCF-7 |
Roscovitine, Proteasome inhibitors: MG-132 and MG-262 |
- |
In vitro |
Proteasome inhibitors did not mimic roscovitine effects for increasing p53 expression |
164
|
| MCF-7 |
ROSC, olomoucine, nocodazole |
- |
In vitro |
Roscovitine-mediated phosphorylation of p53 at Ser-46 stabilizes wild-type p53, leading to apoptosis in cancer cells |
165
|
| MCF-7 |
Roscovitine |
- |
In vitro |
The effect of roscovitine on the proliferation and apoptosis of cancer cells depends on the cell cycle status |
166
|
| MCF-7,HeLaS3 |
Roscovitine |
- |
In vitro |
The outcome of treatment with roscvovitine relies on cell cycle status and drug dose |
167
|
| MCF-7 |
Roscovitine, Olomoucine |
- |
In vitro |
Roscovitine induces apoptosis in MCF-7 cells through the mitochondrial pathway |
99
|
| MCF-7 |
Roscovitine, farnesyltransferase (FPTase) inhibitor: L-774,832 |
- |
In vitro |
Blockade of farnesyl protein transferase in MCF-7 cells enhances the anti-proliferative impact of roscovitine |
168
|
MCF7
HCT116 p53 +/+
HCT116 p53−/− |
Roscovitine, PNUTS-siRNA |
- |
In vitro |
Downregulation of PNUTS increases roscovitine-induced apoptosis in cancer cells |
169
|
| MDA-MB231 |
Roscovitine, olomoucine |
- |
In vitro |
Roscovitine suppresses the proliferation of MDA-MB231 cells through targeting cdk5 |
170
|
| MDA-MB231,MCF10A,MCF-7,T24 |
Roscovitine, Cdk5 shRNA |
Balb/c nude mice |
In vitro/In vivo |
Knockdown of Cdk5 or its inhibition by roscovitine decreased tumor formation and induced cell death in tumor-initiating cells |
171
|
| MCF-7 |
Roscovitine |
- |
In vitro |
Roscovitine promotes apoptosis through the upregulation of p53AIP1 protein in MCF-7 BC cells |
98
|
| MCF-7,MCF-7.0.3,MCF-7.3.28 |
Roscovitine, olomoucine, DRB, etoposide (VP16), staurosporine |
- |
In vitro |
Forced expression of caspase-3 in MCF-7 cells sensitizes these cells to the function of DNA-damaging proteins but not to roscovitine |
172
|
| MCF-7 |
Roscovitine, ornithine decarboxylase inhibitor: DFMO |
- |
In vitro |
Inhibition of ornithine decarboxylase can affect the Roscovitine-mediated apoptosis through the mitochondrial pathway |
173
|
| MCF-7,MDA-MB-231 |
Roscovitine |
- |
In vitro |
Exogenous polyamines play a critical role in the induction of apoptosis or autophagy by roscovitine |
174
|
| MDA-MB-231 (p53 mutated) |
Roscovitine |
SCID (DNA-PKcs deficient) and BALB
athymic nude mice |
In vitro/In vivo |
Roscovitine can sensitize cancer cells against radiation |
175
|
| MDA-MB-231 HMVECs,HMEC-1 |
Roscovitine |
- |
In vitro |
The combination of roscovitine and radiation inhibits the angiogenesis process |
176
|
| MCF-7 |
Roscovitine |
- |
In vitro |
Naturally occurring compounds such as phenol red can enhance the anti-cancer potential of roscovitine |
177
|
| MCF-7 |
Roscovitine, resveratrol |
- |
In vitro |
Resveratrol modulates roscovitine-mediated cell cycle arrest |
178
|
| MDA-MB-231MCF-7,BT 474,SKBr-3,EVSA-T,MDA-MB-435-S |
Roscovitine, TRAIL, Pancaspase inhibitor, (z-VAD.FMK), Cycloheximide (CHX), cFLIP-siRNA
Mcl-1- siRNA, E2F1-siRNA |
- |
In vitro |
Roscovitine enhances the apoptosis of cancer cells by TRAIL |
179
|
| MCF-74T1,BT474,SKBR3 |
Roscovitine, Flavopiridol, ERBB1/ERBB2 inhibitor (Lapatinib), BH3 domain inhibitor (Obatoclax), Si-BCL-2, Si-BCL-X, Si-MCL-1, Si-BAX, Si-BAK |
- |
In vitro |
Modulation of Mcl-1 expression by roscovitine sensitizes BC cells to BAX/BAK-dependent mitochondrial apoptosis |
180
|
| MCF-7 |
Seliciclib, doxorubicin |
Nude (nu/nu) mice |
In vitro/In vivo |
Roscovitine increases the antitumor effect of doxorubicin |
100
|
| MCF7,MDA-MB-436, HCC1806,HCC1569,ZR75-1, UACC812, MCF-7 |
Roscovitine, doxorubicin, roscovitine, meriolin 5, Cdk2 inhibitor: CVT-313 |
Nulliparous mice |
In vitro
In vitro/In vivo |
Roscovitine in combination with doxorubicin synergistically enhanced apoptosis in LMW-E-expressing cells
Silencing of Cdk2 enhances apoptosis in LMW-E overexpressing cell lines |
181,182
|
| MCF-7,Ac1 |
Roscovitine, aromatase inhibitors: letrozole and exemestane anastrozole |
- |
In vitro |
Roscovitine reverses resistance to letrozole due to LMW-E expression |
183
|
| HMEC,MCF-10A,76NE6,76NF2V,MDA-MB-157,MDA-MB-231,HCC1806,MCF-7,ZR75-1,T47D,MDAMB-468, HCT116 p53 +/+, HCT116 p53−/−, HEK-293T |
Roscovitine, doxorubicine |
Balb/c Nu/nu mice |
In vitro/In vivo |
Sequential combination treatment of roscovitine and doxorubicin synergistically inhibits p53-mutant TNBC cells |
184
|
MCF-7
SKBr3
BT474 |
Roscovitine, HER2/neu siRNA, trastuzumab |
Nude mice |
In vitro/In vivo |
HER2-mediated signalling affects LMW cyclin E expression and cell cycle |
185
|
| MCF7,A549,H460,SkBr3,H1650,H292,H358 |
Seliciclib, AG1478, trastuzumab |
(Nu/nu) mice |
In vitro/In vivo |
Roscovitine and the anti-HER-2 antibody synergistically suppress tumor growth |
186
|
| MCF-7, MCF7 MDR |
Roscovitive, Sangivamycin, Caspase-6 inhibitor (Z-VEID-FMK), Caspase-3/7 inhibitor I |
- |
In vitro |
Roscovitine induces apoptosis in drug-resistant P-gp–overexpressing BC cells |
187
|
| MCF-7,BT-20,SKBr-3 |
Roscovitine, DRB, tamoxifen, 4-HOT |
- |
In vitro |
Roscovitine can inhibit the activating phosphorylation of ER-α and exhibit synergistic effects in combination with anti-estrogen therapy |
89
|
| MCF-7,MCF-7-E6,BT-20, SKBr-3 |
Roscovitine, tamoxifen, Staurosporine, DRB, Estradiol (E2), 4-HOT |
- |
In vitro |
Tamoxifen increases the anti-proliferative function of roscovitine |
188-190
|
| MCF7-LTLTca, CF7-TamR |
Roscovitine |
Nude mice |
In vitro/In vivo |
Roscovitine suppresses tumor growth in hormone therapy-resistant tumors |
114
|
| T47D(T47D-YB,T47D PR-null,T47D wt PR-B,T47D S345A PR),MCF-7,COS-1,BT-474 cells |
Roscovitine, MAPK inhibitor (U0126) |
- |
In vitro |
Blockade of CDK2 by roscovitine inhibits the interaction of PR/cyclin D1 with DNA and reduces the expression of HSPB8 |
191
|
Note. SiRNA: Small interfering RNA; TRAIL: Tumor necrosis factor-related apoptosis-inducing ligand; PNUTS: Phosphatase nuclear targeting subunit; LMW: Low molecular weight; TNBC: Triple-negative breast cancer; CDK: Cyclin-dependent kinase; MAPK: Mitogen-activated protein kinase; ER: Estrogen receptor; PR: Partial response; DRB: 5,6-dichlorobenzimidazole 1-b-D-ribofuranoside; 4-HOT: 4-hydroxytamoxifen; P-gp: P-glycoprotein
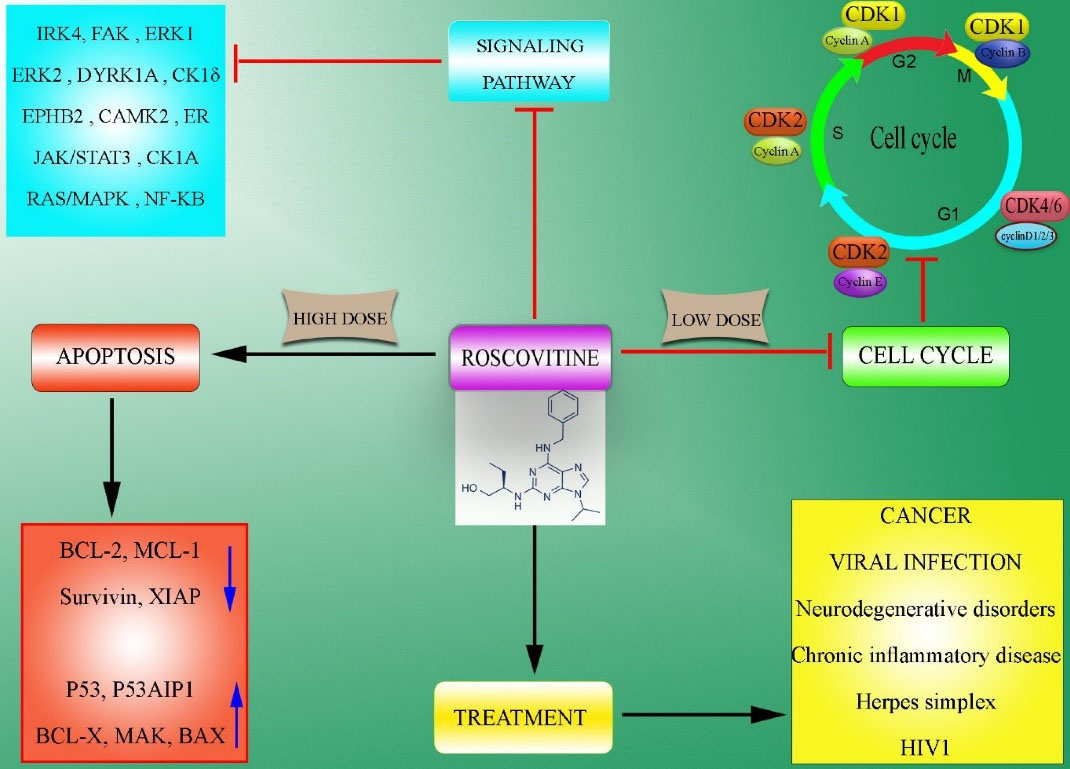
Figure 5.
Action Mechanisms of Roscovitine. Note. Roscovitine can arrest the cell cycle in G0, G1, S, or G2/M phases in the dose-, time-, and cell-type-dependent manner by suppressing CDKs. Although Roscovitine suppresses cell cycle progression at low doses through direct inhibition of CDKs, it induces apoptosis at high doses, which is partly through the downregulation of anti-apoptotic factors such as BCL-2, MCL-1, Survivin, and XIAP, as well as the upregulation of pro-apoptotic factors such as P53, BAK, and BAX. It has also been demonstrated that Roscovitine can modulate several signaling pathways such as Ras-MAPK, NF-κβ, p53, estrogen receptor, JAK-STAT, and the like. In addition to cancer diseases, Roscovitine can be an effective therapeutic option for the treatment of several pathological conditions such as viral infections, glomerulonephritis, chronic inflammatory disease, neurodegenerative disorders, HIV, and Herpes simplex infections. BCL-2: B-cell lymphoma 2; MCL-1: Myeloid cell leukemia 1; XIAP: X-linked inhibitor of apoptosis protein; BAK: BCL2-antagonist/killer 1; BAX: Bcl-2-associated X protein, MAPK: Mitogen-activated protein kinase; NF-κβ: Nuclear factor kappa light chain enhancer of activated B cells; JAK-STAT: Janus kinase/signal transducers and activators of transcription.
.
Action Mechanisms of Roscovitine. Note. Roscovitine can arrest the cell cycle in G0, G1, S, or G2/M phases in the dose-, time-, and cell-type-dependent manner by suppressing CDKs. Although Roscovitine suppresses cell cycle progression at low doses through direct inhibition of CDKs, it induces apoptosis at high doses, which is partly through the downregulation of anti-apoptotic factors such as BCL-2, MCL-1, Survivin, and XIAP, as well as the upregulation of pro-apoptotic factors such as P53, BAK, and BAX. It has also been demonstrated that Roscovitine can modulate several signaling pathways such as Ras-MAPK, NF-κβ, p53, estrogen receptor, JAK-STAT, and the like. In addition to cancer diseases, Roscovitine can be an effective therapeutic option for the treatment of several pathological conditions such as viral infections, glomerulonephritis, chronic inflammatory disease, neurodegenerative disorders, HIV, and Herpes simplex infections. BCL-2: B-cell lymphoma 2; MCL-1: Myeloid cell leukemia 1; XIAP: X-linked inhibitor of apoptosis protein; BAK: BCL2-antagonist/killer 1; BAX: Bcl-2-associated X protein, MAPK: Mitogen-activated protein kinase; NF-κβ: Nuclear factor kappa light chain enhancer of activated B cells; JAK-STAT: Janus kinase/signal transducers and activators of transcription.
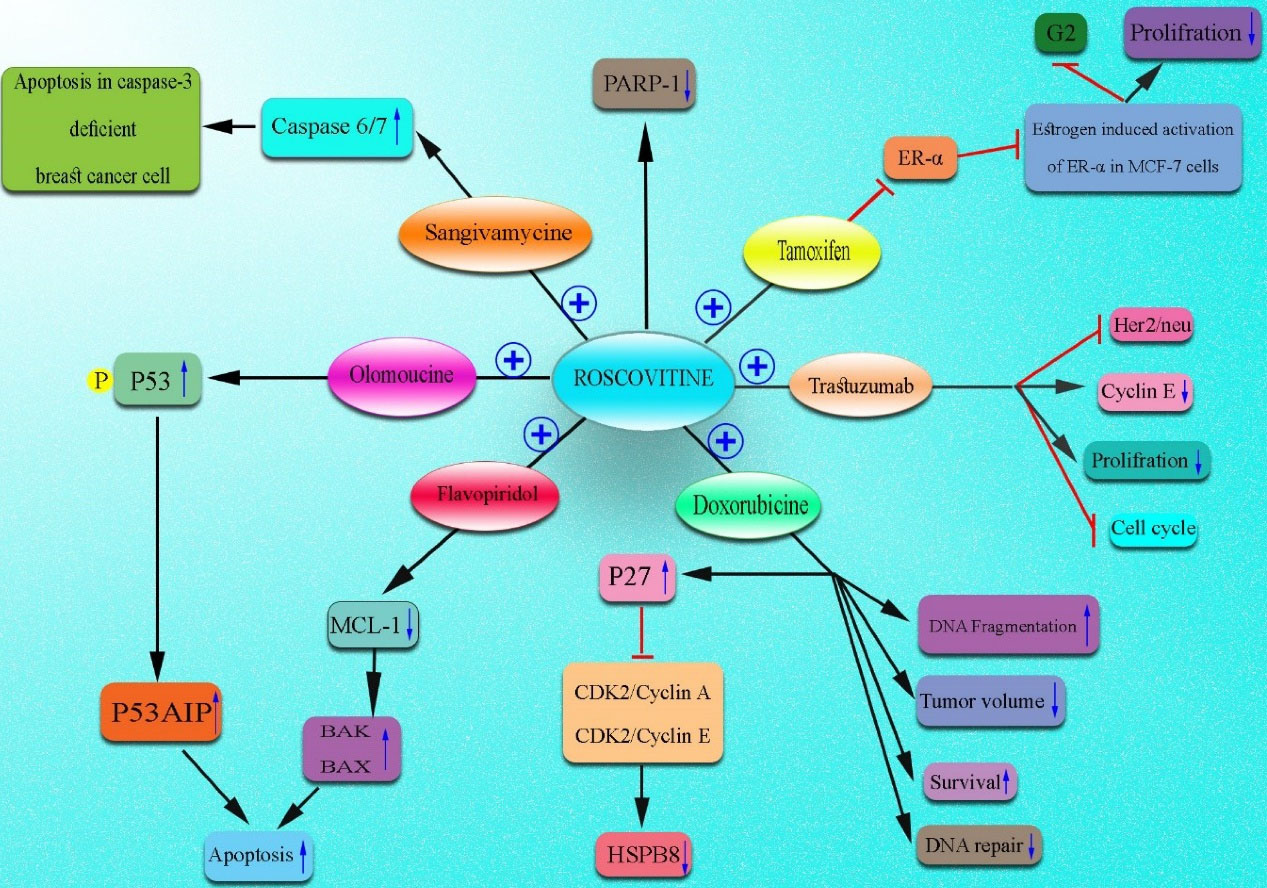
Figure 6.
Combination of Roscovitine With Other Therapeutics for Breast Cancer Therapy. Note. Several anti-cancer therapeutics have been used in combination with Roscovitine to achieve synergistic ameliorative effects. It has been demonstrated that Roscovitine in combination with olomoucine induces apoptosis in BC cells through the upregulation of the p53AIP1 protein. A combination of Roscovitine and sangivamycin also induces apoptosis in breast cancer cells via the upregulation of caspase 6/7. Additionally, Roscovitine and Doxorubicin synergistically exerted anti-cancer effects. Tamoxifen is another therapeutic which has been successfully employed in combination with Roscovitine. Trastuzumab and Flavopiridol also represented synergism when combined with Roscovitine for the treatment of BC. p53AIP1: p53-regulated apoptosis-inducing protein 1; BC: Breast cancer.
.
Combination of Roscovitine With Other Therapeutics for Breast Cancer Therapy. Note. Several anti-cancer therapeutics have been used in combination with Roscovitine to achieve synergistic ameliorative effects. It has been demonstrated that Roscovitine in combination with olomoucine induces apoptosis in BC cells through the upregulation of the p53AIP1 protein. A combination of Roscovitine and sangivamycin also induces apoptosis in breast cancer cells via the upregulation of caspase 6/7. Additionally, Roscovitine and Doxorubicin synergistically exerted anti-cancer effects. Tamoxifen is another therapeutic which has been successfully employed in combination with Roscovitine. Trastuzumab and Flavopiridol also represented synergism when combined with Roscovitine for the treatment of BC. p53AIP1: p53-regulated apoptosis-inducing protein 1; BC: Breast cancer.
Conclusion
Although several outstanding discoveries regarding the treatment of BC resulted in the low mortality of patients with BC during the last three decades, its treatment is yet one of the main health-related challenges. Currently available anti-BC therapeutics are usually safe, well-tolerated, and efficient; however, cancer recurrence following the emergence of drug-resistant cancer cells often leads to the appearance of more advanced, progressive, and metastatic tumors. Therefore, the identification of novel effective therapeutic approaches is a critical issue on this topic. Accordingly, new therapeutic targets need to be identified to overcome current treatment limitations.192 Cancer cells show rapid proliferation, implying the presence of dysregulated cell cycle modulators. CDKs are the main molecules involved in the regulation of the cell cycle and transcription process. Thus, several pharmacological inhibitors of CDKs have been developed in recent years as a novel promising treatment strategy against several cancer types, including BC. However, the unknown precise action mechanism of CDK inhibitors has limited their use, alone or in combination with other anti-cancer therapeutics, in the clinic.193 Pan CDK inhibitors are the most important CDK inhibitors that enable the suppression of various CDKs, including the CDK1, 2, 5, 7, and 9.194 Several in vitro studies have reported that they can exert cell cycle arrest and apoptosis in a wide variety of BC cell lines. Although preclinical studies using Pan CDK inhibitors, alone or in combination with other therapeutics, were associated with potent breast tumor growth arrest, clinical trials were not hopeful. However, it should be noted that some trials were performed to evaluate the efficacy of these CDK inhibitors in cancer therapy (https://www.clinicaltrials.gov/). Hence, further trials are crucial to assign the safety and efficacy of Pan CDK inhibitors in patients with BC. Based on the results of preclinical and clinical studies, it seems that combination therapies with Pan CDK inhibitors may be more effective compared to monotherapy; nonetheless, the identification of the most effective combined strategy is a crucial issue, which should be addressed in future studies. Another proposal for future studies is the design, development, and evaluation of new derivatives of Pan CDK inhibitors.195,196
Ethics statement
Not applicable.
Disclosure of funding source
This study was supported by grant from Tabriz University of Medical Sciences (grant number: 69964).
Conflict of interests declaration
All authors declare no conflict of interests.
Data availability statement
Not applicable.
Consent for publication
Not applicable.
References
- Siegel R, Naishadham D, Jemal A. Cancer statistics, 2013. CA Cancer J Clin 2013; 63(1):11-30. doi: 10.3322/caac.21166 [Crossref] [ Google Scholar]
- Adams GP, Weiner LM. Monoclonal antibody therapy of cancer. Nat Biotechnol 2005; 23(9):1147-57. doi: 10.1038/nbt1137 [Crossref] [ Google Scholar]
- Schwob E. [Nobel Prize in Medicine 2001: the universal key to cell division]. Bull Cancer 2001;88(10):937. [French].
- Malumbres M, Barbacid M. Mammalian cyclin-dependent kinases. Trends Biochem Sci 2005; 30(11):630-41. doi: 10.1016/j.tibs.2005.09.005 [Crossref] [ Google Scholar]
- Satyanarayana A, Kaldis P. Mammalian cell-cycle regulation: several Cdks, numerous cyclins and diverse compensatory mechanisms. Oncogene 2009; 28(33):2925-39. doi: 10.1038/onc.2009.170 [Crossref] [ Google Scholar]
- Bhattacharya S, Ray RM, Johnson LR. Cyclin-dependent kinases regulate apoptosis of intestinal epithelial cells. Apoptosis 2014; 19(3):451-66. doi: 10.1007/s10495-013-0942-3 [Crossref] [ Google Scholar]
- Canavese M, Santo L, Raje N. Cyclin dependent kinases in cancer: potential for therapeutic intervention. Cancer Biol Ther 2012; 13(7):451-7. doi: 10.4161/cbt.19589 [Crossref] [ Google Scholar]
- Whittaker SR, Mallinger A, Workman P, Clarke PA. Inhibitors of cyclin-dependent kinases as cancer therapeutics. Pharmacol Ther 2017; 173:83-105. doi: 10.1016/j.pharmthera.2017.02.008 [Crossref] [ Google Scholar]
- DiPippo AJ, Patel NK, Barnett CM. Cyclin-dependent kinase inhibitors for the treatment of breast cancer: past, present, and future. Pharmacotherapy 2016; 36(6):652-67. doi: 10.1002/phar.1756 [Crossref] [ Google Scholar]
- Syn NL, Lim PL, Kong LR, Wang L, Wong AL, Lim CM. Pan-CDK inhibition augments cisplatin lethality in nasopharyngeal carcinoma cell lines and xenograft models. Signal Transduct Target Ther 2018; 3:9. doi: 10.1038/s41392-018-0010-0 [Crossref] [ Google Scholar]
- Siegel RL, Miller KD, Jemal A. Cancer statistics, 2016. CA Cancer J Clin 2016; 66(1):7-30. doi: 10.3322/caac.21332 [Crossref] [ Google Scholar]
- Ferzoco RM, Ruddy KJ. The epidemiology of male breast cancer. Curr Oncol Rep 2016; 18(1):1. doi: 10.1007/s11912-015-0487-4 [Crossref] [ Google Scholar]
- Plevritis SK, Munoz D, Kurian AW, Stout NK, Alagoz O, Near AM. Association of screening and treatment with breast cancer mortality by molecular subtype in US women, 2000-2012. JAMA 2018; 319(2):154-64. doi: 10.1001/jama.2017.19130 [Crossref] [ Google Scholar]
- DeSantis CE, Bray F, Ferlay J, Lortet-Tieulent J, Anderson BO, Jemal A. International variation in female breast cancer incidence and mortality rates. Cancer Epidemiol Biomarkers Prev 2015; 24(10):1495-506. doi: 10.1158/1055-9965.epi-15-0535 [Crossref] [ Google Scholar]
- Vahabi M. Breast cancer screening methods: a review of the evidence. Health Care Women Int 2003; 24(9):773-93. doi: 10.1080/07399330390229957 [Crossref] [ Google Scholar]
- Liu Z, Zhang XS, Zhang S. Breast tumor subgroups reveal diverse clinical prognostic power. Sci Rep 2014; 4:4002. doi: 10.1038/srep04002 [Crossref] [ Google Scholar]
- Dai X, Li T, Bai Z, Yang Y, Liu X, Zhan J. Breast cancer intrinsic subtype classification, clinical use and future trends. Am J Cancer Res 2015; 5(10):2929-43. [ Google Scholar]
- Hayes EL, Lewis-Wambi JS. Mechanisms of endocrine resistance in breast cancer: an overview of the proposed roles of noncoding RNA. Breast Cancer Res 2015; 17:40. doi: 10.1186/s13058-015-0542-y [Crossref] [ Google Scholar]
- Massarweh S, Schiff R. Unraveling the mechanisms of endocrine resistance in breast cancer: new therapeutic opportunities. Clin Cancer Res 2007; 13(7):1950-4. doi: 10.1158/1078-0432.ccr-06-2540 [Crossref] [ Google Scholar]
- Hudis CA. Trastuzumab--mechanism of action and use in clinical practice. N Engl J Med 2007; 357(1):39-51. doi: 10.1056/NEJMra043186 [Crossref] [ Google Scholar]
- Barry M, Ho A, Morrow M. The evolving role of partial breast irradiation in early-stage breast cancer. Ann Surg Oncol 2013; 20(8):2534-40. doi: 10.1245/s10434-013-2923-8 [Crossref] [ Google Scholar]
- Ovcaricek T, Frkovic SG, Matos E, Mozina B, Borstnar S. Triple negative breast cancer-prognostic factors and survival. Radiol Oncol 2011; 45(1):46-52. doi: 10.2478/v10019-010-0054-4 [Crossref] [ Google Scholar]
- Crescenzi E, Palumbo G, Brady HJ. Roscovitine modulates DNA repair and senescence: implications for combination chemotherapy. Clin Cancer Res 2005; 11(22):8158-71. doi: 10.1158/1078-0432.ccr-05-1042 [Crossref] [ Google Scholar]
- Lapenna S, Giordano A. Cell cycle kinases as therapeutic targets for cancer. Nat Rev Drug Discov 2009; 8(7):547-66. doi: 10.1038/nrd2907 [Crossref] [ Google Scholar]
- Cicenas J, Valius M. The CDK inhibitors in cancer research and therapy. J Cancer Res Clin Oncol 2011; 137(10):1409-18. doi: 10.1007/s00432-011-1039-4 [Crossref] [ Google Scholar]
- Cicenas J, Kalyan K, Sorokinas A, Jatulyte A, Valiunas D, Kaupinis A. Highlights of the latest advances in research on CDK inhibitors. Cancers (Basel) 2014; 6(4):2224-42. doi: 10.3390/cancers6042224 [Crossref] [ Google Scholar]
- Gray N, Détivaud L, Doerig C, Meijer L. ATP-site directed inhibitors of cyclin-dependent kinases. Curr Med Chem 1999; 6(9):859-75. [ Google Scholar]
- Abaza MS, Bahman AM, Al-Attiyah RJ. Roscovitine synergizes with conventional chemo-therapeutic drugs to induce efficient apoptosis of human colorectal cancer cells. World J Gastroenterol 2008; 14(33):5162-75. doi: 10.3748/wjg.14.5162 [Crossref] [ Google Scholar]
- Fry DW, Harvey PJ, Keller PR, Elliott WL, Meade M, Trachet E. Specific inhibition of cyclin-dependent kinase 4/6 by PD 0332991 and associated antitumor activity in human tumor xenografts. Mol Cancer Ther 2004; 3(11):1427-38. [ Google Scholar]
- Ehab M, Elbaz M. Profile of palbociclib in the treatment of metastatic breast cancer. Breast Cancer (Dove Med Press) 2016; 8:83-91. doi: 10.2147/bctt.s83146 [Crossref] [ Google Scholar]
- Altenburg JD, Farag SS. The potential role of PD0332991 (Palbociclib) in the treatment of multiple myeloma. Expert Opin Investig Drugs 2015; 24(2):261-71. doi: 10.1517/13543784.2015.993753 [Crossref] [ Google Scholar]
- Li C, Qi L, Bellail AC, Hao C, Liu T. PD-0332991 induces G1 arrest of colorectal carcinoma cells through inhibition of the cyclin-dependent kinase-6 and retinoblastoma protein axis. Oncol Lett 2014; 7(5):1673-8. doi: 10.3892/ol.2014.1957 [Crossref] [ Google Scholar]
- Huang S, Ye H, Guo W, Dong X, Wu N, Zhang X. CDK4/6 inhibitor suppresses gastric cancer with CDKN2A mutation. Int J Clin Exp Med 2015; 8(7):11692-700. [ Google Scholar]
- Perez M, Muñoz-Galván S, Jiménez-García MP, Marín JJ, Carnero A. Efficacy of CDK4 inhibition against sarcomas depends on their levels of CDK4 and p16ink4 mRNA. Oncotarget 2015; 6(38):40557-74. doi: 10.18632/oncotarget.5829 [Crossref] [ Google Scholar]
- Rihani A, Vandesompele J, Speleman F, Van Maerken T. Inhibition of CDK4/6 as a novel therapeutic option for neuroblastoma. Cancer Cell Int 2015; 15:76. doi: 10.1186/s12935-015-0224-y [Crossref] [ Google Scholar]
- Schröder LB, McDonald KL. CDK4/6 inhibitor PD0332991 in glioblastoma treatment: does it have a future?. Front Oncol 2015; 5:259. doi: 10.3389/fonc.2015.00259 [Crossref] [ Google Scholar]
- Sathe A, Koshy N, Schmid SC, Thalgott M, Schwarzenböck SM, Krause BJ. CDK4/6 inhibition controls proliferation of bladder cancer and transcription of RB1. J Urol 2016; 195(3):771-9. doi: 10.1016/j.juro.2015.08.082 [Crossref] [ Google Scholar]
- Murphy CG, Dickler MN. The role of CDK4/6 inhibition in breast cancer. Oncologist 2015; 20(5):483-90. doi: 10.1634/theoncologist.2014-0443 [Crossref] [ Google Scholar]
- Finn RS, Dering J, Conklin D, Kalous O, Cohen DJ, Desai AJ. PD 0332991, a selective cyclin D kinase 4/6 inhibitor, preferentially inhibits proliferation of luminal estrogen receptor-positive human breast cancer cell lines in vitro. Breast Cancer Res 2009; 11(5):R77. doi: 10.1186/bcr2419 [Crossref] [ Google Scholar]
- Dean JL, McClendon AK, Hickey TE, Butler LM, Tilley WD, Witkiewicz AK. Therapeutic response to CDK4/6 inhibition in breast cancer defined by ex vivo analyses of human tumors. Cell Cycle 2012; 11(14):2756-61. doi: 10.4161/cc.21195 [Crossref] [ Google Scholar]
- Finn RS, Martin M, Rugo HS, Jones S, Im SA, Gelmon K. Palbociclib and letrozole in advanced breast cancer. N Engl J Med 2016; 375(20):1925-36. doi: 10.1056/NEJMoa1607303 [Crossref] [ Google Scholar]
- Infante JR, Cassier PA, Gerecitano JF, Witteveen PO, Chugh R, Ribrag V. A phase I study of the cyclin-dependent kinase 4/6 inhibitor ribociclib (LEE011) in patients with advanced solid tumors and lymphomas. Clin Cancer Res 2016; 22(23):5696-705. doi: 10.1158/1078-0432.ccr-16-1248 [Crossref] [ Google Scholar]
- Goel S, Wang Q, Watt AC, Tolaney SM, Dillon DA, Li W. Overcoming therapeutic resistance in HER2-positive breast cancers with CDK4/6 inhibitors. Cancer Cell 2016; 29(3):255-69. doi: 10.1016/j.ccell.2016.02.006 [Crossref] [ Google Scholar]
- Hortobagyi GN, Stemmer SM, Burris HA, Yap YS, Sonke GS, Paluch-Shimon S. Ribociclib as first-line therapy for HR-positive, advanced breast cancer. N Engl J Med 2016; 375(18):1738-48. doi: 10.1056/NEJMoa1609709 [Crossref] [ Google Scholar]
- Hortobagyi GN, Stemmer SM, Burris HA, Yap YS, Sonke GS, Paluch-Shimon S. Updated results from MONALEESA-2, a phase III trial of first-line ribociclib plus letrozole versus placebo plus letrozole in hormone receptor-positive, HER2-negative advanced breast cancer. Ann Oncol 2018; 29(7):1541-7. doi: 10.1093/annonc/mdy155 [Crossref] [ Google Scholar]
- Slamon DJ, Neven P, Chia S, Fasching PA, De Laurentiis M, Im SA. Phase III randomized study of ribociclib and fulvestrant in hormone receptor-positive, human epidermal growth factor receptor 2-negative advanced breast cancer: MONALEESA-3. J Clin Oncol 2018; 36(24):2465-72. doi: 10.1200/jco.2018.78.9909 [Crossref] [ Google Scholar]
- Tripathy D, Im SA, Colleoni M, Franke F, Bardia A, Harbeck N. Ribociclib plus endocrine therapy for premenopausal women with hormone-receptor-positive, advanced breast cancer (MONALEESA-7): a randomised phase 3 trial. Lancet Oncol 2018; 19(7):904-15. doi: 10.1016/s1470-2045(18)30292-4 [Crossref] [ Google Scholar]
- Lallena MJ, Boehnke K, Torres R, Hermoso A, Amat J, Calsina B. In-vitro characterization of abemaciclib pharmacology in ER + breast cancer cell lines. Cancer Res 2015; 75(15 Suppl):3101. doi: 10.1158/1538-7445.am2015-3101 [Crossref] [ Google Scholar]
- Gelbert LM, Cai S, Lin X, Sanchez-Martinez C, Del Prado M, Lallena MJ. Preclinical characterization of the CDK4/6 inhibitor LY2835219: in-vivo cell cycle-dependent/independent anti-tumor activities alone/in combination with gemcitabine. Invest New Drugs 2014; 32(5):825-37. doi: 10.1007/s10637-014-0120-7 [Crossref] [ Google Scholar]
- Yadav V, Burke TF, Huber L, Van Horn RD, Zhang Y, Buchanan SG. The CDK4/6 inhibitor LY2835219 overcomes vemurafenib resistance resulting from MAPK reactivation and cyclin D1 upregulation. Mol Cancer Ther 2014; 13(10):2253-63. doi: 10.1158/1535-7163.mct-14-0257 [Crossref] [ Google Scholar]
- Raub TJ, Wishart GN, Kulanthaivel P, Staton BA, Ajamie RT, Sawada GA. Brain exposure of two selective dual CDK4 and CDK6 inhibitors and the antitumor activity of CDK4 and CDK6 inhibition in combination with temozolomide in an intracranial glioblastoma xenograft. Drug Metab Dispos 2015; 43(9):1360-71. doi: 10.1124/dmd.114.062745 [Crossref] [ Google Scholar]
- Ku BM, Yi SY, Koh J, Bae YH, Sun JM, Lee SH. The CDK4/6 inhibitor LY2835219 has potent activity in combination with mTOR inhibitor in head and neck squamous cell carcinoma. Oncotarget 2016; 7(12):14803-13. doi: 10.18632/oncotarget.7543 [Crossref] [ Google Scholar]
- Patnaik A, Rosen LS, Tolaney SM, Tolcher AW, Goldman JW, Gandhi L. Efficacy and safety of abemaciclib, an inhibitor of CDK4 and CDK6, for patients with breast cancer, non-small cell lung cancer, and other solid tumors. Cancer Discov 2016; 6(7):740-53. doi: 10.1158/2159-8290.cd-16-0095 [Crossref] [ Google Scholar]
- Wu T, Chen Z, To KKW, Fang X, Wang F, Cheng B. Effect of abemaciclib (LY2835219) on enhancement of chemotherapeutic agents in ABCB1 and ABCG2 overexpressing cells in vitro and in vivo. Biochem Pharmacol 2017; 124:29-42. doi: 10.1016/j.bcp.2016.10.015 [Crossref] [ Google Scholar]
- Senderowicz AM. The cell cycle as a target for cancer therapy: basic and clinical findings with the small molecule inhibitors flavopiridol and UCN-01. Oncologist 2002; 7 Suppl 3:12-9. doi: 10.1634/theoncologist.7-suppl_3-12 [Crossref] [ Google Scholar]
- Zhai S, Senderowicz AM, Sausville EA, Figg WD. Flavopiridol, a novel cyclin-dependent kinase inhibitor, in clinical development. Ann Pharmacother 2002; 36(5):905-11. doi: 10.1345/aph.1A162 [Crossref] [ Google Scholar]
- Chao SH, Price DH. Flavopiridol inactivates P-TEFb and blocks most RNA polymerase II transcription in vivo. J Biol Chem 2001; 276(34):31793-9. doi: 10.1074/jbc.M102306200 [Crossref] [ Google Scholar]
- Sekine C, Sugihara T, Miyake S, Hirai H, Yoshida M, Miyasaka N. Successful treatment of animal models of rheumatoid arthritis with small-molecule cyclin-dependent kinase inhibitors. J Immunol 2008; 180(3):1954-61. doi: 10.4049/jimmunol.180.3.1954 [Crossref] [ Google Scholar]
- Parry D, Guzi T, Shanahan F, Davis N, Prabhavalkar D, Wiswell D. Dinaciclib (SCH 727965), a novel and potent cyclin-dependent kinase inhibitor. Mol Cancer Ther 2010; 9(8):2344-53. doi: 10.1158/1535-7163.mct-10-0324 [Crossref] [ Google Scholar]
- Abdullah C, Wang X, Becker D. Expression analysis and molecular targeting of cyclin-dependent kinases in advanced melanoma: functional analysis and molecular targeting of cyclin-dependent kinase family members in advanced melanoma. Cell Cycle 2011; 10(6):977-88. doi: 10.4161/cc.10.6.15079 [Crossref] [ Google Scholar]
- Feldmann G, Mishra A, Bisht S, Karikari C, Garrido-Laguna I, Rasheed Z. Cyclin-dependent kinase inhibitor dinaciclib (SCH727965) inhibits pancreatic cancer growth and progression in murine xenograft models. Cancer Biol Ther 2011; 12(7):598-609. doi: 10.4161/cbt.12.7.16475 [Crossref] [ Google Scholar]
- Fu W, Ma L, Chu B, Wang X, Bui MM, Gemmer J. The cyclin-dependent kinase inhibitor SCH 727965 (dinacliclib) induces the apoptosis of osteosarcoma cells. Mol Cancer Ther 2011; 10(6):1018-27. doi: 10.1158/1535-7163.mct-11-0167 [Crossref] [ Google Scholar]
- Desai BM, Villanueva J, Nguyen TT, Lioni M, Xiao M, Kong J. The anti-melanoma activity of dinaciclib, a cyclin-dependent kinase inhibitor, is dependent on p53 signaling. PLoS One 2013; 8(3):e59588. doi: 10.1371/journal.pone.0059588 [Crossref] [ Google Scholar]
- Johnson AJ, Yeh YY, Smith LL, Wagner AJ, Hessler J, Gupta S. The novel cyclin-dependent kinase inhibitor dinaciclib (SCH727965) promotes apoptosis and abrogates microenvironmental cytokine protection in chronic lymphocytic leukemia cells. Leukemia 2012; 26(12):2554-7. doi: 10.1038/leu.2012.144 [Crossref] [ Google Scholar]
- Ma H, Seebacher NA, Hornicek FJ. Cyclin-dependent kinase 9 (CDK9) is a novel prognostic marker and therapeutic target in osteosarcoma. EBioMedicine 2019; 39:182-193. doi: 10.1016/j.ebiom.2018.12.022 [Crossref] [ Google Scholar]
- Yang J, Hu S, Shangguan J. Dinaciclib prolongs survival in the LSL-KrasG12D/+; LSL-Trp53R172H/+; Pdx-1-Cre (KPC) transgenic murine models of pancreatic ductal adenocarcinoma. Am J Transl Res 2020; 12(3):1031-1043. [ Google Scholar]
- Horiuchi D, Kusdra L, Huskey NE, Chandriani S, Lenburg ME, Gonzalez-Angulo AM. MYC pathway activation in triple-negative breast cancer is synthetic lethal with CDK inhibition. J Exp Med 2012; 209(4):679-96. doi: 10.1084/jem.20111512 [Crossref] [ Google Scholar]
- Rajput S, Khera N, Guo Z, Hoog J, Li S, Ma CX. Inhibition of cyclin dependent kinase 9 by dinaciclib suppresses cyclin B1 expression and tumor growth in triple negative breast cancer. Oncotarget 2016; 7(35):56864-56875. doi: 10.18632/oncotarget.10870 [Crossref] [ Google Scholar]
- Flynn J, Jones J, Johnson AJ, Andritsos L, Maddocks K, Jaglowski S. Dinaciclib is a novel cyclin-dependent kinase inhibitor with significant clinical activity in relapsed and refractory chronic lymphocytic leukemia. Leukemia 2015; 29(7):1524-9. doi: 10.1038/leu.2015.31 [Crossref] [ Google Scholar]
- Gregory GP, Hogg SJ, Kats LM, Vidacs E, Baker AJ, Gilan O. CDK9 inhibition by dinaciclib potently suppresses Mcl-1 to induce durable apoptotic responses in aggressive MYC-driven B-cell lymphoma in vivo. Leukemia 2015; 29(6):1437-41. doi: 10.1038/leu.2015.10 [Crossref] [ Google Scholar]
- Kumar SK, LaPlant B, Chng WJ, Zonder J, Callander N, Fonseca R. Dinaciclib, a novel CDK inhibitor, demonstrates encouraging single-agent activity in patients with relapsed multiple myeloma. Blood 2015; 125(3):443-8. doi: 10.1182/blood-2014-05-573741 [Crossref] [ Google Scholar]
- Mita MM, Joy AA, Mita A, Sankhala K, Jou YM, Zhang D. Randomized phase II trial of the cyclin-dependent kinase inhibitor dinaciclib (MK-7965) versus capecitabine in patients with advanced breast cancer. Clin Breast Cancer 2014; 14(3):169-76. doi: 10.1016/j.clbc.2013.10.016 [Crossref] [ Google Scholar]
- Stephenson JJ, Nemunaitis J, Joy AA, Martin JC, Jou YM, Zhang D. Randomized phase 2 study of the cyclin-dependent kinase inhibitor dinaciclib (MK-7965) versus erlotinib in patients with non-small cell lung cancer. Lung Cancer 2014; 83(2):219-23. doi: 10.1016/j.lungcan.2013.11.020 [Crossref] [ Google Scholar]
- Bose P, Simmons GL, Grant S. Cyclin-dependent kinase inhibitor therapy for hematologic malignancies. Expert Opin Investig Drugs 2013; 22(6):723-38. doi: 10.1517/13543784.2013.789859 [Crossref] [ Google Scholar]
- Phillips HS, Kharbanda S, Chen R, Forrest WF, Soriano RH, Wu TD. Molecular subclasses of high-grade glioma predict prognosis, delineate a pattern of disease progression, and resemble stages in neurogenesis. Cancer Cell 2006; 9(3):157-73. doi: 10.1016/j.ccr.2006.02.019 [Crossref] [ Google Scholar]
- Ducray F, Idbaih A, de Reyniès A, Bièche I, Thillet J, Mokhtari K. Anaplastic oligodendrogliomas with 1p19q codeletion have a proneural gene expression profile. Mol Cancer 2008; 7:41. doi: 10.1186/1476-4598-7-41 [Crossref] [ Google Scholar]
- Cooper LA, Gutman DA, Long Q, Johnson BA, Cholleti SR, Kurc T. The proneural molecular signature is enriched in oligodendrogliomas and predicts improved survival among diffuse gliomas. PLoS One 2010; 5(9):e12548. doi: 10.1371/journal.pone.0012548 [Crossref] [ Google Scholar]
- Kim SH, Ezhilarasan R, Phillips E, Gallego-Perez D, Sparks A, Taylor D. Serine/threonine kinase MLK4 determines mesenchymal identity in glioma stem cells in an NF-κB-dependent manner. Cancer Cell 2016; 29(2):201-13. doi: 10.1016/j.ccell.2016.01.005 [Crossref] [ Google Scholar]
- Tong WG, Chen R, Plunkett W, Siegel D, Sinha R, Harvey RD. Phase I and pharmacologic study of SNS-032, a potent and selective Cdk2, 7, and 9 inhibitor, in patients with advanced chronic lymphocytic leukemia and multiple myeloma. J Clin Oncol 2010; 28(18):3015-22. doi: 10.1200/jco.2009.26.1347 [Crossref] [ Google Scholar]
- Chen R, Wierda WG, Chubb S, Hawtin RE, Fox JA, Keating MJ. Mechanism of action of SNS-032, a novel cyclin-dependent kinase inhibitor, in chronic lymphocytic leukemia. Blood 2009; 113(19):4637-45. doi: 10.1182/blood-2008-12-190256 [Crossref] [ Google Scholar]
- Xie G, Tang H, Wu S, Chen J, Liu J, Liao C. The cyclin-dependent kinase inhibitor SNS-032 induces apoptosis in breast cancer cells via depletion of Mcl-1 and X-linked inhibitor of apoptosis protein and displays antitumor activity in vivo. Int J Oncol 2014; 45(2):804-12. doi: 10.3892/ijo.2014.2467 [Crossref] [ Google Scholar]
- Meijer L, Borgne A, Mulner O, Chong JP, Blow JJ, Inagaki N. Biochemical and cellular effects of roscovitine, a potent and selective inhibitor of the cyclin-dependent kinases cdc2, cdk2 and cdk5. Eur J Biochem 1997; 243(1-2):527-36. doi: 10.1111/j.1432-1033.1997.t01-2-00527.x [Crossref] [ Google Scholar]
- De Azevedo WF, Leclerc S, Meijer L, Havlicek L, Strnad M, Kim SH. Inhibition of cyclin-dependent kinases by purine analogues: crystal structure of human cdk2 complexed with roscovitine. Eur J Biochem 1997; 243(1-2):518-26. doi: 10.1111/j.1432-1033.1997.0518a.x [Crossref] [ Google Scholar]
- Nutley BP, Raynaud FI, Wilson SC, Fischer PM, Hayes A, Goddard PM. Metabolism and pharmacokinetics of the cyclin-dependent kinase inhibitor R-roscovitine in the mouse. Mol Cancer Ther 2005; 4(1):125-39. [ Google Scholar]
- Gray NS, Wodicka L, Thunnissen AM, Norman TC, Kwon S, Espinoza FH. Exploiting chemical libraries, structure, and genomics in the search for kinase inhibitors. Science 1998; 281(5376):533-8. doi: 10.1126/science.281.5376.533 [Crossref] [ Google Scholar]
- Bain J, McLauchlan H, Elliott M, Cohen P. The specificities of protein kinase inhibitors: an update. Biochem J 2003; 371(Pt 1):199-204. doi: 10.1042/bj20021535 [Crossref] [ Google Scholar]
- Bach S, Knockaert M, Reinhardt J, Lozach O, Schmitt S, Baratte B. Roscovitine targets, protein kinases and pyridoxal kinase. J Biol Chem 2005; 280(35):31208-19. doi: 10.1074/jbc.M500806200 [Crossref] [ Google Scholar]
- Mohapatra S, Chu B, Wei S, Djeu J, Epling-Burnette PK, Loughran T. Roscovitine inhibits STAT5 activity and induces apoptosis in the human leukemia virus type 1-transformed cell line MT-2. Cancer Res 2003; 63(23):8523-30. [ Google Scholar]
- Węsierska-Gądek J, Gritsch D, Zulehner N, Komina O, Maurer M. Roscovitine, a selective CDK inhibitor, reduces the basal and estrogen-induced phosphorylation of ER-α in human ER-positive breast cancer cells. J Cell Biochem 2011; 112(3):761-72. doi: 10.1002/jcb.23004 [Crossref] [ Google Scholar]
- Paprskárová M, Krystof V, Jorda R, Dzubák P, Hajdúch M, Wesierska-Gadek J. Functional p53 in cells contributes to the anticancer effect of the cyclin-dependent kinase inhibitor roscovitine. J Cell Biochem 2009; 107(3):428-37. doi: 10.1002/jcb.22139 [Crossref] [ Google Scholar]
- Dey A, Wong ET, Cheok CF, Tergaonkar V, Lane DP. R-roscovitine simultaneously targets both the p53 and NF-kappaB pathways and causes potentiation of apoptosis: implications in cancer therapy. Cell Death Differ 2008; 15(2):263-73. doi: 10.1038/sj.cdd.4402257 [Crossref] [ Google Scholar]
- Whittaker SR, Walton MI, Garrett MD, Workman P. The cyclin-dependent kinase inhibitor CYC202 (R-roscovitine) inhibits retinoblastoma protein phosphorylation, causes loss of cyclin D1, and activates the mitogen-activated protein kinase pathway. Cancer Res 2004; 64(1):262-72. doi: 10.1158/0008-5472.can-03-0110 [Crossref] [ Google Scholar]
- Kim EH, Kim SU, Shin DY, Choi KS. Roscovitine sensitizes glioma cells to TRAIL-mediated apoptosis by downregulation of survivin and XIAP. Oncogene 2004; 23(2):446-56. doi: 10.1038/sj.onc.1207025 [Crossref] [ Google Scholar]
- MacCallum DE, Melville J, Frame S, Watt K, Anderson S, Gianella-Borradori A. Seliciclib (CYC202, R-roscovitine) induces cell death in multiple myeloma cells by inhibition of RNA polymerase II-dependent transcription and down-regulation of Mcl-1. Cancer Res 2005; 65(12):5399-407. doi: 10.1158/0008-5472.can-05-0233 [Crossref] [ Google Scholar]
- Coker-Gurkan A, Arısan ED, Obakan P, Ozfiliz P, Kose B, Bickici G. Roscovitine-treated HeLa cells finalize autophagy later than apoptosis by downregulating Bcl-2. Mol Med Rep 2015; 11(3):1968-74. doi: 10.3892/mmr.2014.2902 [Crossref] [ Google Scholar]
- Mihara M, Shintani S, Kiyota A, Matsumura T, Wong DT. Cyclin-dependent kinase inhibitor (roscovitine) suppresses growth and induces apoptosis by regulating Bcl-x in head and neck squamous cell carcinoma cells. Int J Oncol 2002; 21(1):95-101. [ Google Scholar]
- Wesierska-Gadek J, Wandl S, Kramer MP, Pickem C, Krystof V, Hajek SB. Roscovitine up-regulates p53 protein and induces apoptosis in human HeLaS3 cervix carcinoma cells. J Cell Biochem 2008; 105(5):1161-71. doi: 10.1002/jcb.21903 [Crossref] [ Google Scholar]
- Wesierska-Gadek J, Gueorguieva M, Horky M. Roscovitine-induced up-regulation of p53AIP1 protein precedes the onset of apoptosis in human MCF-7 breast cancer cells. Mol Cancer Ther 2005; 4(1):113-24. [ Google Scholar]
- Wesierska-Gadek J, Gueorguieva M, Wojciechowski J, Horky M. Cell cycle arrest induced in human breast cancer cells by cyclin-dependent kinase inhibitors: a comparison of the effects exerted by roscovitine and olomoucine. Pol J Pharmacol 2004; 56(5):635-41. [ Google Scholar]
- Appleyard MV, O’Neill MA, Murray KE, Paulin FE, Bray SE, Kernohan NM. Seliciclib (CYC202, R-roscovitine) enhances the antitumor effect of doxorubicin in vivo in a breast cancer xenograft model. Int J Cancer 2009; 124(2):465-72. doi: 10.1002/ijc.23938 [Crossref] [ Google Scholar]
- Zhang T, Jiang T, Zhang F, Li C, Zhou YA, Zhu YF. Involvement of p21Waf1/Cip1 cleavage during roscovitine-induced apoptosis in non-small cell lung cancer cells. Oncol Rep 2010; 23(1):239-45. [ Google Scholar]
- Arısan ED, Coker A, Palavan-Ünsal N. Polyamine depletion enhances the roscovitine-induced apoptosis through the activation of mitochondria in HCT116 colon carcinoma cells. Amino Acids 2012; 42(2-3):655-65. doi: 10.1007/s00726-011-1040-x [Crossref] [ Google Scholar]
- Cicenas J, Kalyan K, Sorokinas A, Stankunas E, Levy J, Meskinyte I. Roscovitine in cancer and other diseases. Ann Transl Med 2015; 3(10):135. doi: 10.3978/j.issn.2305-5839.2015.03.61 [Crossref] [ Google Scholar]
- Ozfiliz-Kilbas P, Sarikaya B, Obakan-Yerlikaya P, Coker-Gurkan A, Arısan ED, Temizci B. Cyclin-dependent kinase inhibitors, roscovitine and purvalanol, induce apoptosis and autophagy related to unfolded protein response in HeLa cervical cancer cells. Mol Biol Rep 2018; 45(5):815-28. doi: 10.1007/s11033-018-4222-8 [Crossref] [ Google Scholar]
- Rossi AG, Sawatzky DA, Walker A, Ward C, Sheldrake TA, Riley NA. Cyclin-dependent kinase inhibitors enhance the resolution of inflammation by promoting inflammatory cell apoptosis. Nat Med 2006; 12(9):1056-64. doi: 10.1038/nm1468 [Crossref] [ Google Scholar]
- McClue SJ, Blake D, Clarke R, Cowan A, Cummings L, Fischer PM. In vitro and in vivo antitumor properties of the cyclin dependent kinase inhibitor CYC202 (R-roscovitine). Int J Cancer 2002; 102(5):463-8. doi: 10.1002/ijc.10738 [Crossref] [ Google Scholar]
- Raynaud FI, Whittaker SR, Fischer PM, McClue S, Walton MI, Barrie SE. In vitro and in vivo pharmacokinetic-pharmacodynamic relationships for the trisubstituted aminopurine cyclin-dependent kinase inhibitors olomoucine, bohemine and CYC202. Clin Cancer Res 2005; 11(13):4875-87. doi: 10.1158/1078-0432.ccr-04-2264 [Crossref] [ Google Scholar]
- Tirado OM, Mateo-Lozano S, Notario V. Roscovitine is an effective inducer of apoptosis of Ewing’s sarcoma family tumor cells in vitro and in vivo. Cancer Res 2005; 65(20):9320-7. doi: 10.1158/0008-5472.can-05-1276 [Crossref] [ Google Scholar]
- Payton M, Chung G, Yakowec P, Wong A, Powers D, Xiong L. Discovery and evaluation of dual CDK1 and CDK2 inhibitors. Cancer Res 2006; 66(8):4299-308. doi: 10.1158/0008-5472.can-05-2507 [Crossref] [ Google Scholar]
- Iurisci I, Filipski E, Reinhardt J, Bach S, Gianella-Borradori A, Iacobelli S. Improved tumor control through circadian clock induction by seliciclib, a cyclin-dependent kinase inhibitor. Cancer Res 2006; 66(22):10720-8. doi: 10.1158/0008-5472.can-06-2086 [Crossref] [ Google Scholar]
- Abal M, Bras-Goncalves R, Judde JG, Fsihi H, De Cremoux P, Louvard D. Enhanced sensitivity to irinotecan by Cdk1 inhibition in the p53-deficient HT29 human colon cancer cell line. Oncogene 2004; 23(9):1737-44. doi: 10.1038/sj.onc.1207299 [Crossref] [ Google Scholar]
- Molinsky J, Klanova M, Koc M, Beranova L, Andera L, Ludvikova Z. Roscovitine sensitizes leukemia and lymphoma cells to tumor necrosis factor-related apoptosis-inducing ligand-induced apoptosis. Leuk Lymphoma 2013; 54(2):372-80. doi: 10.3109/10428194.2012.710331 [Crossref] [ Google Scholar]
- Appleyard MV, O’Neill MA, Murray KE, Paulin FE, Bray SE, Kernohan NM. Seliciclib (CYC202, R-roscovitine) enhances the antitumor effect of doxorubicin in vivo in a breast cancer xenograft model. Int J Cancer 2009; 124(2):465-72. doi: 10.1002/ijc.23938 [Crossref] [ Google Scholar]
- Nair BC, Vallabhaneni S, Tekmal RR, Vadlamudi RK. Roscovitine confers tumor suppressive effect on therapy-resistant breast tumor cells. Breast Cancer Res 2011; 13(3):R80. doi: 10.1186/bcr2929 [Crossref] [ Google Scholar]
- Lichota A, Gwozdzinski K. Anticancer activity of natural compounds from plant and marine environment. Int J Mol Sci 2018; 19(11):3533. doi: 10.3390/ijms19113533 [Crossref] [ Google Scholar]
- Schwartz GK, Farsi K, Maslak P, Kelsen DP, Spriggs D. Potentiation of apoptosis by flavopiridol in mitomycin-C-treated gastric and breast cancer cells. Clin Cancer Res 1997; 3(9):1467-72. [ Google Scholar]
- Motwani M, Delohery TM, Schwartz GK. Sequential dependent enhancement of caspase activation and apoptosis by flavopiridol on paclitaxel-treated human gastric and breast cancer cells. Clin Cancer Res 1999; 5(7):1876-83. [ Google Scholar]
- Li Y, Bhuiyan M, Alhasan S, Senderowicz AM, Sarkar FH. Induction of apoptosis and inhibition of c-erbB-2 in breast cancer cells by flavopiridol. Clin Cancer Res 2000; 6(1):223-9. [ Google Scholar]
- Carlson B, Lahusen T, Singh S, Loaiza-Perez A, Worland PJ, Pestell R. Down-regulation of cyclin D1 by transcriptional repression in MCF-7 human breast carcinoma cells induced by flavopiridol. Cancer Res 1999; 59(18):4634-41. [ Google Scholar]
- Nahta R, Iglehart JD, Kempkes B, Schmidt EV. Rate-limiting effects of cyclin D1 in transformation by ErbB2 predicts synergy between herceptin and flavopiridol. Cancer Res 2002; 62(8):2267-71. [ Google Scholar]
- Nahta R, Trent S, Yang C, Schmidt EV. Epidermal growth factor receptor expression is a candidate target of the synergistic combination of trastuzumab and flavopiridol in breast cancer. Cancer Res 2003; 63(13):3626-31. [ Google Scholar]
- Wu K, Wang C, D’Amico M, Lee RJ, Albanese C, Pestell RG. Flavopiridol and trastuzumab synergistically inhibit proliferation of breast cancer cells: association with selective cooperative inhibition of cyclin D1-dependent kinase and Akt signaling pathways. Mol Cancer Ther 2002; 1(9):695-706. [ Google Scholar]
- Wall NR, O’Connor DS, Plescia J, Pommier Y, Altieri DC. Suppression of survivin phosphorylation on Thr34 by flavopiridol enhances tumor cell apoptosis. Cancer Res 2003; 63(1):230-5. [ Google Scholar]
- Wittmann S, Bali P, Donapaty S, Nimmanapalli R, Guo F, Yamaguchi H. Flavopiridol down-regulates antiapoptotic proteins and sensitizes human breast cancer cells to epothilone B-induced apoptosis. Cancer Res 2003; 63(1):93-9. [ Google Scholar]
- Najmi S, Korah R, Chandra R, Abdellatif M, Wieder R. Flavopiridol blocks integrin-mediated survival in dormant breast cancer cells. Clin Cancer Res 2005; 11(5):2038-46. doi: 10.1158/1078-0432.ccr-04-1083 [Crossref] [ Google Scholar]
- Ali S, El-Rayes BF, Aranha O, Sarkar FH, Philip PA. Sequence dependent potentiation of gemcitabine by flavopiridol in human breast cancer cells. Breast Cancer Res Treat 2005; 90(1):25-31. doi: 10.1007/s10549-004-2179-x [Crossref] [ Google Scholar]
- Mayer F, Mueller S, Malenke E, Kuczyk M, Hartmann JT, Bokemeyer C. Induction of apoptosis by flavopiridol unrelated to cell cycle arrest in germ cell tumour derived cell lines. Invest New Drugs 2005; 23(3):205-11. doi: 10.1007/s10637-005-6728-x [Crossref] [ Google Scholar]
- Palacios C, Yerbes R, López-Rivas A. Flavopiridol induces cellular FLICE-inhibitory protein degradation by the proteasome and promotes TRAIL-induced early signaling and apoptosis in breast tumor cells. Cancer Res 2006; 66(17):8858-69. doi: 10.1158/0008-5472.can-06-0808 [Crossref] [ Google Scholar]
- Fandy TE, Ross DD, Gore SD, Srivastava RK. Flavopiridol synergizes TRAIL cytotoxicity by downregulation of FLIPL. Cancer Chemother Pharmacol 2007; 60(3):313-9. doi: 10.1007/s00280-006-0381-8 [Crossref] [ Google Scholar]
- Mitchell C, Park MA, Zhang G, Yacoub A, Curiel DT, Fisher PB. Extrinsic pathway- and cathepsin-dependent induction of mitochondrial dysfunction are essential for synergistic flavopiridol and vorinostat lethality in breast cancer cells. Mol Cancer Ther 2007; 6(12 Pt 1):3101-12. doi: 10.1158/1535-7163.mct-07-0561 [Crossref] [ Google Scholar]
- Fornier MN, Rathkopf D, Shah M, Patil S, O’Reilly E, Tse AN. Phase I dose-finding study of weekly docetaxel followed by flavopiridol for patients with advanced solid tumors. Clin Cancer Res 2007; 13(19):5841-6. doi: 10.1158/1078-0432.ccr-07-1218 [Crossref] [ Google Scholar]
- Lamb R, Lehn S, Rogerson L, Clarke RB, Landberg G. Cell cycle regulators cyclin D1 and CDK4/6 have estrogen receptor-dependent divergent functions in breast cancer migration and stem cell-like activity. Cell Cycle 2013; 12(15):2384-94. doi: 10.4161/cc.25403 [Crossref] [ Google Scholar]
- Nagaria TS, Williams JL, Leduc C, Squire JA, Greer PA, Sangrar W. Flavopiridol synergizes with sorafenib to induce cytotoxicity and potentiate antitumorigenic activity in EGFR/HER-2 and mutant RAS/RAF breast cancer model systems. Neoplasia 2013; 15(8):939-51. doi: 10.1593/neo.13804 [Crossref] [ Google Scholar]
- Hicks M, Hu Q, Macrae E, DeWille J. JUNB promotes the survival of flavopiridol treated human breast cancer cells. Biochem Biophys Res Commun 2014; 450(1):19-24. doi: 10.1016/j.bbrc.2014.05.057 [Crossref] [ Google Scholar]
- Brenner S, Riha J, Giessrigl B, Thalhammer T, Grusch M, Krupitza G. The effect of organic anion-transporting polypeptides 1B1, 1B3 and 2B1 on the antitumor activity of flavopiridol in breast cancer cells. Int J Oncol 2015; 46(1):324-32. doi: 10.3892/ijo.2014.2731 [Crossref] [ Google Scholar]
- Shao X, Gao D, Wang Y, Jin F, Wu Q, Liu H. Application of metabolomics to investigate the antitumor mechanism of flavopiridol in MCF-7 breast cancer cells. J Chromatogr B Analyt Technol Biomed Life Sci 2016; 1025:40-7. doi: 10.1016/j.jchromb.2016.05.009 [Crossref] [ Google Scholar]
- Okada Y, Kato S, Sakamoto Y, Oishi T, Ishioka C. Synthetic lethal interaction of CDK inhibition and autophagy inhibition in human solid cancer cell lines. Oncol Rep 2017; 38(1):31-42. doi: 10.3892/or.2017.5684 [Crossref] [ Google Scholar]
- Wang S, Wang K, Wang H, Han J, Sun H. Autophagy is essential for flavopiridol-induced cytotoxicity against MCF-7 breast cancer cells. Mol Med Rep 2017; 16(6):9715-20. doi: 10.3892/mmr.2017.7815 [Crossref] [ Google Scholar]
- Erol A, Acikgoz E, Guven U, Duzagac F, Turkkani A, Colcimen N. Ribosome biogenesis mediates antitumor activity of flavopiridol in CD44( + )/CD24(-) breast cancer stem cells. Oncol Lett 2017; 14(6):6433-40. doi: 10.3892/ol.2017.7029 [Crossref] [ Google Scholar]
- Mitri Z, Karakas C, Wei C, Briones B, Simmons H, Ibrahim N. A phase 1 study with dose expansion of the CDK inhibitor dinaciclib (SCH 727965) in combination with epirubicin in patients with metastatic triple negative breast cancer. Invest New Drugs 2015; 33(4):890-4. doi: 10.1007/s10637-015-0244-4 [Crossref] [ Google Scholar]
- Scott GK, Chu D, Kaur R, Malato J, Rothschild DE, Frazier K. ERpS294 is a biomarker of ligand or mutational ERα activation and a breast cancer target for CDK2 inhibition. Oncotarget 2017; 8(48):83432-45. doi: 10.18632/oncotarget.12735 [Crossref] [ Google Scholar]
- Johnson SF, Cruz C, Greifenberg AK, Dust S, Stover DG, Chi D. CDK12 inhibition reverses de novo and acquired PARP inhibitor resistance in BRCA wild-type and mutated models of triple-negative breast cancer. Cell Rep 2016; 17(9):2367-81. doi: 10.1016/j.celrep.2016.10.077 [Crossref] [ Google Scholar]
- Alexander A, Karakas C, Chen X, Carey JP, Yi M, Bondy M. Cyclin E overexpression as a biomarker for combination treatment strategies in inflammatory breast cancer. Oncotarget 2017; 8(9):14897-911. doi: 10.18632/oncotarget.14689 [Crossref] [ Google Scholar]
- Jansen VM, Bhola NE, Bauer JA, Formisano L, Lee KM, Hutchinson KE. Kinome-wide RNA interference screen reveals a role for PDK1 in acquired resistance to CDK4/6 inhibition in ER-positive breast cancer. Cancer Res 2017; 77(9):2488-99. doi: 10.1158/0008-5472.can-16-2653 [Crossref] [ Google Scholar]
- Doostan I, Karakas C, Kohansal M, Low KH, Ellis MJ, Olson JA Jr. Cytoplasmic cyclin E mediates resistance to aromatase inhibitors in breast cancer. Clin Cancer Res 2017; 23(23):7288-300. doi: 10.1158/1078-0432.ccr-17-1544 [Crossref] [ Google Scholar]
- Zhu Y, Liu Y, Zhang C, Chu J, Wu Y, Li Y. Tamoxifen-resistant breast cancer cells are resistant to DNA-damaging chemotherapy because of upregulated BARD1 and BRCA1. Nat Commun 2018; 9(1):1595. doi: 10.1038/s41467-018-03951-0 [Crossref] [ Google Scholar]
- Urasaki Y, Fiscus RR, Le TT. Detection of the cell cycle-regulated negative feedback phosphorylation of mitogen-activated protein kinases in breast carcinoma using nanofluidic proteomics. Sci Rep 2018; 8(1):9991. doi: 10.1038/s41598-018-28335-8 [Crossref] [ Google Scholar]
- Chen X, Low KH, Alexander A, Jiang Y, Karakas C, Hess KR. Cyclin E overexpression sensitizes triple-negative breast cancer to Wee1 kinase inhibition. Clin Cancer Res 2018; 24(24):6594-610. doi: 10.1158/1078-0432.ccr-18-1446 [Crossref] [ Google Scholar]
- Carey JPW, Karakas C, Bui T, Chen X, Vijayaraghavan S, Zhao Y. Synthetic lethality of PARP inhibitors in combination with MYC blockade is independent of BRCA status in triple-negative breast cancer. Cancer Res 2018; 78(3):742-57. doi: 10.1158/0008-5472.can-17-1494 [Crossref] [ Google Scholar]
- Nie L, Wei Y, Zhang F, Hsu YH, Chan LC, Xia W. CDK2-mediated site-specific phosphorylation of EZH2 drives and maintains triple-negative breast cancer. Nat Commun 2019; 10(1):5114. doi: 10.1038/s41467-019-13105-5 [Crossref] [ Google Scholar]
- Ma Y, Freeman SN, Cress WD. E2F4 deficiency promotes drug-induced apoptosis. Cancer Biol Ther 2004; 3(12):1262-9. doi: 10.4161/cbt.3.12.1239 [Crossref] [ Google Scholar]
- Ma Y, Cress WD. Transcriptional upregulation of p57 (Kip2) by the cyclin-dependent kinase inhibitor BMS-387032 is E2F dependent and serves as a negative feedback loop limiting cytotoxicity. Oncogene 2007; 26(24):3532-40. doi: 10.1038/sj.onc.1210143 [Crossref] [ Google Scholar]
- Held JM, Britton DJ, Scott GK, Lee EL, Schilling B, Baldwin MA. Ligand binding promotes CDK-dependent phosphorylation of ER-alpha on hinge serine 294 but inhibits ligand-independent phosphorylation of serine 305. Mol Cancer Res 2012; 10(8):1120-32. doi: 10.1158/1541-7786.mcr-12-0099 [Crossref] [ Google Scholar]
- Xie G, Tang H, Wu S, Chen J, Liu J, Liao C. The cyclin-dependent kinase inhibitor SNS-032 induces apoptosis in breast cancer cells via depletion of Mcl-1 and X-linked inhibitor of apoptosis protein and displays antitumor activity in vivo. Int J Oncol 2014; 45(2):804-12. doi: 10.3892/ijo.2014.2467 [Crossref] [ Google Scholar]
- Cihalova D, Staud F, Ceckova M. Interactions of cyclin-dependent kinase inhibitors AT-7519, flavopiridol and SNS-032 with ABCB1, ABCG2 and ABCC1 transporters and their potential to overcome multidrug resistance in vitro. Cancer Chemother Pharmacol 2015; 76(1):105-16. doi: 10.1007/s00280-015-2772-1 [Crossref] [ Google Scholar]
- Yanagi T, Shi R, Aza-Blanc P, Reed JC, Matsuzawa S. PCTAIRE1-knockdown sensitizes cancer cells to TNF family cytokines. PLoS One 2015; 10(3):e0119404. doi: 10.1371/journal.pone.0119404 [Crossref] [ Google Scholar]
- Villerbu N, Gaben AM, Redeuilh G, Mester J. Cellular effects of purvalanol A: a specific inhibitor of cyclin-dependent kinase activities. Int J Cancer 2002; 97(6):761-9. doi: 10.1002/ijc.10125 [Crossref] [ Google Scholar]
- Greenberg VL, Zimmer SG. Paclitaxel induces the phosphorylation of the eukaryotic translation initiation factor 4E-binding protein 1 through a Cdk1-dependent mechanism. Oncogene 2005; 24(30):4851-60. doi: 10.1038/sj.onc.1208624 [Crossref] [ Google Scholar]
- Hofman J, Ahmadimoghaddam D, Hahnova L, Pavek P, Ceckova M, Staud F. Olomoucine II and purvalanol A inhibit ABCG2 transporter in vitro and in situ and synergistically potentiate cytostatic effect of mitoxantrone. Pharmacol Res 2012; 65(3):312-9. doi: 10.1016/j.phrs.2011.11.017 [Crossref] [ Google Scholar]
- Obakan P, Arısan ED, Özfiliz P, Çoker-Gürkan A, Palavan-Ünsal N. Purvalanol A is a strong apoptotic inducer via activating polyamine catabolic pathway in MCF-7 estrogen receptor positive breast cancer cells. Mol Biol Rep 2014; 41(1):145-54. doi: 10.1007/s11033-013-2847-1 [Crossref] [ Google Scholar]
- Mgbonyebi OP, Russo J, Russo IH. Roscovitine inhibits the proliferative activity of immortal and neoplastic human breast epithelial cells. Anticancer Res 1998; 18(2A):751-5. [ Google Scholar]
- Mgbonyebi OP, Russo J, Russo IH. Roscovitine induces cell death and morphological changes indicative of apoptosis in MDA-MB-231 breast cancer cells. Cancer Res 1999; 59(8):1903-10. [ Google Scholar]
- Wojciechowski J, Horky M, Gueorguieva M, Węsierska-Gądek J. Rapid onset of nucleolar disintegration preceding cell cycle arrest in roscovitine-induced apoptosis of human MCF-7 breast cancer cells. Int J Cancer 2003; 106(4):486-95. doi: 10.1002/ijc.11290 [Crossref] [ Google Scholar]
- Ranftler C, Gueorguieva M, Węsierska-Gądek J. Prevention of p53 degradation in human MCF-7 cells by proteasome inhibitors does not mimic the action of roscovitine. Ann N Y Acad Sci 2006; 1090:234-44. doi: 10.1196/annals.1378.026 [Crossref] [ Google Scholar]
- Węsierska-Gądek J, Schmitz ML, Ranftler C. Roscovitine-activated HIP2 kinase induces phosphorylation of wt p53 at Ser-46 in human MCF-7 breast cancer cells. J Cell Biochem 2007; 100(4):865-74. doi: 10.1002/jcb.21211 [Crossref] [ Google Scholar]
- Maurer M, Komina O, Węsierska-Gądek J. Roscovitine differentially affects asynchronously growing and synchronized human MCF-7 breast cancer cells. Ann N Y Acad Sci 2009; 1171:250-6. doi: 10.1111/j.1749-6632.2009.04717.x [Crossref] [ Google Scholar]
- Węsierska-Gądek J, Borza A, Komina O, Maurer M. Impact of roscovitine, a selective CDK inhibitor, on cancer cells: bi-functionality increases its therapeutic potential. Acta Biochim Pol 2009; 56(3):495-501. [ Google Scholar]
- Węsierska-Gądek J, Maurer M, Schmid G. Inhibition of farnesyl protein transferase sensitizes human MCF-7 breast cancer cells to roscovitine-mediated cell cycle arrest. J Cell Biochem 2007; 102(3):736-47. doi: 10.1002/jcb.21325 [Crossref] [ Google Scholar]
- De Leon G, Cavino M, D’Angelo M, Krucher NA. PNUTS knockdown potentiates the apoptotic effect of roscovitine in breast and colon cancer cells. Int J Oncol 2010; 36(5):1269-75. doi: 10.3892/ijo_00000611 [Crossref] [ Google Scholar]
- Goodyear S, Sharma MC. Roscovitine regulates invasive breast cancer cell (MDA-MB231) proliferation and survival through cell cycle regulatory protein cdk5. Exp Mol Pathol 2007; 82(1):25-32. doi: 10.1016/j.yexmp.2006.09.002 [Crossref] [ Google Scholar]
- Mandl MM, Zhang S, Ulrich M, Schmoeckel E, Mayr D, Vollmar AM. Inhibition of Cdk5 induces cell death of tumor-initiating cells. Br J Cancer 2017; 116(7):912-22. doi: 10.1038/bjc.2017.39 [Crossref] [ Google Scholar]
- Węsierska-Gądek J, Hackl S, Zulehner N, Maurer M, Komina O. Reconstitution of human MCF-7 breast cancer cells with caspase-3 does not sensitize them to action of CDK inhibitors. J Cell Biochem 2011; 112(1):273-88. doi: 10.1002/jcb.22918 [Crossref] [ Google Scholar]
- Arısan ED, Obakan P, Coker A, Palavan-Ünsal N. Inhibition of ornithine decarboxylase alters the roscovitine-induced mitochondrial-mediated apoptosis in MCF-7 breast cancer cells. Mol Med Rep 2012; 5(5):1323-9. doi: 10.3892/mmr.2012.786 [Crossref] [ Google Scholar]
- Arısan ED, Akkoç Y, Akyüz KG, Kerman EM, Obakan P, Çoker-Gürkan A. Polyamines modulate the roscovitine-induced cell death switch decision autophagy vs apoptosis in MCF-7 and MDA-MB-231 breast cancer cells. Mol Med Rep 2015; 11(6):4532-40. doi: 10.3892/mmr.2015.3303 [Crossref] [ Google Scholar]
- Maggiorella L, Deutsch E, Frascogna V, Chavaudra N, Jeanson L, Milliat F. Enhancement of radiation response by roscovitine in human breast carcinoma in vitro and in vivo. Cancer Res 2003; 63(10):2513-7. [ Google Scholar]
- Maggiorella L, Aubel C, Haton C, Milliat F, Connault E, Opolon P. Cooperative effect of roscovitine and irradiation targets angiogenesis and induces vascular destabilization in human breast carcinoma. Cell Prolif 2009; 42(1):38-48. doi: 10.1111/j.1365-2184.2008.00570.x [Crossref] [ Google Scholar]
- Węsierska-Gądek J, Schreiner T, Maurer M, Waringer A, Ranftler C. Phenol red in the culture medium strongly affects the susceptibility of human MCF-7 cells to roscovitine. Cell Mol Biol Lett 2007; 12(2):280-93. doi: 10.2478/s11658-007-0002-5 [Crossref] [ Google Scholar]
- Węsierska-Gądek J, Kramer MP, Maurer M. Resveratrol modulates roscovitine-mediated cell cycle arrest of human MCF-7 breast cancer cells. Food Chem Toxicol 2008; 46(4):1327-33. doi: 10.1016/j.fct.2007.09.004 [Crossref] [ Google Scholar]
- Ortiz-Ferrón G, Yerbes R, Eramo A, López-Pérez AI, De Maria R, López-Rivas A. Roscovitine sensitizes breast cancer cells to TRAIL-induced apoptosis through a pleiotropic mechanism. Cell Res 2008; 18(6):664-76. doi: 10.1038/cr.2008.54 [Crossref] [ Google Scholar]
- Mitchell C, Yacoub A, Hossein H, Martin AP, Bareford MD, Eulitt P. Inhibition of MCL-1 in breast cancer cells promotes cell death in vitro and in vivo. Cancer Biol Ther 2010; 10(9):903-17. doi: 10.4161/cbt.10.9.13273 [Crossref] [ Google Scholar]
- Nanos-Webb A, Jabbour NA, Multani AS, Wingate H, Oumata N, Galons H. Targeting low molecular weight cyclin E (LMW-E) in breast cancer. Breast Cancer Res Treat 2012; 132(2):575-88. doi: 10.1007/s10549-011-1638-4 [Crossref] [ Google Scholar]
- Akli S, Van Pelt CS, Bui T, Meijer L, Keyomarsi K. Cdk2 is required for breast cancer mediated by the low-molecular-weight isoform of cyclin E. Cancer Res 2011; 71(9):3377-86. doi: 10.1158/0008-5472.can-10-4086 [Crossref] [ Google Scholar]
- Akli S, Bui T, Wingate H, Biernacka A, Moulder S, Tucker SL. Low-molecular-weight cyclin E can bypass letrozole-induced G1 arrest in human breast cancer cells and tumors. Clin Cancer Res 2010; 16(4):1179-90. doi: 10.1158/1078-0432.ccr-09-1787 [Crossref] [ Google Scholar]
- Jabbour-Leung NA, Chen X, Bui T, Jiang Y, Yang D, Vijayaraghavan S. Sequential combination therapy of CDK inhibition and doxorubicin is synthetically lethal in p53-mutant triple-negative breast cancer. Mol Cancer Ther 2016; 15(4):593-607. doi: 10.1158/1535-7163.mct-15-0519 [Crossref] [ Google Scholar]
- Mittendorf EA, Liu Y, Tucker SL, McKenzie T, Qiao N, Akli S. A novel interaction between HER2/neu and cyclin E in breast cancer. Oncogene 2010; 29(27):3896-907. doi: 10.1038/onc.2010.151 [Crossref] [ Google Scholar]
- Fleming IN, Hogben M, Frame S, McClue SJ, Green SR. Synergistic inhibition of ErbB signaling by combined treatment with seliciclib and ErbB-targeting agents. Clin Cancer Res 2008; 14(13):4326-35. doi: 10.1158/1078-0432.ccr-07-4633 [Crossref] [ Google Scholar]
- Cappellini A, Chiarini F, Ognibene A, McCubrey JA, Martelli AM. The cyclin-dependent kinase inhibitor roscovitine and the nucleoside analog sangivamycin induce apoptosis in caspase-3 deficient breast cancer cells independent of caspase mediated P-glycoprotein cleavage: implications for therapy of drug resistant breast cancers. Cell Cycle 2009; 8(9):1421-5. doi: 10.4161/cc.8.9.8323 [Crossref] [ Google Scholar]
- Zulehner N, Maurer M, Wesierska-Gadek J. Effect of anti-estrogen combined with roscovitine, a selective CDK inhibitor, on human breast cancer cells differing in expression of ER. J Exp Ther Oncol 2011; 9(1):17-25. [ Google Scholar]
- Gritsch D, Maurer M, Zulehner N, Wesierska-Gadek J. Tamoxifen enhances the anti-proliferative effect of roscovitine, a selective cyclin-dependent kinase inhibitor, on human ER-positive human breast cancer cells. J Exp Ther Oncol 2011; 9(1):37-45. [ Google Scholar]
- Węsierska-Gądek J, Gritsch D, Zulehner N, Komina O, Maurer M. Interference with ER-α enhances the therapeutic efficacy of the selective CDK inhibitor roscovitine towards ER-positive breast cancer cells. J Cell Biochem 2011; 112(4):1103-17. doi: 10.1002/jcb.23024 [Crossref] [ Google Scholar]
- Dressing GE, Knutson TP, Schiewer MJ, Daniel AR, Hagan CR, Diep CH. Progesterone receptor-cyclin D1 complexes induce cell cycle-dependent transcriptional programs in breast cancer cells. Mol Endocrinol 2014; 28(4):442-57. doi: 10.1210/me.2013-1196 [Crossref] [ Google Scholar]
- Ehab M, Elbaz M. Profile of palbociclib in the treatment of metastatic breast cancer. Breast Cancer (Dove Med Press) 2016; 8:83-91. doi: 10.2147/bctt.s83146 [Crossref] [ Google Scholar]
- Whittaker SR, Mallinger A, Workman P, Clarke PA. Inhibitors of cyclin-dependent kinases as cancer therapeutics. Pharmacol Ther 2017; 173:83-105. doi: 10.1016/j.pharmthera.2017.02.008 [Crossref] [ Google Scholar]
- Heptinstall AB, Adiyasa I, Cano C, Hardcastle IR. Recent advances in CDK inhibitors for cancer therapy. Future Med Chem 2018; 10(11):1369-88. doi: 10.4155/fmc-2017-0246 [Crossref] [ Google Scholar]
- Malínková V, Vylíčil J, Kryštof V. Cyclin-dependent kinase inhibitors for cancer therapy: a patent review (2009-2014). Expert Opin Ther Pat 2015; 25(9):953-70. doi: 10.1517/13543776.2015.1045414 [Crossref] [ Google Scholar]
- Coxon CR, Anscombe E, Harnor SJ, Martin MP, Carbain B, Golding BT. Cyclin-dependent kinase (CDK) inhibitors: structure-activity relationships and insights into the CDK-2 selectivity of 6-substituted 2-arylaminopurines. J Med Chem 2017; 60(5):1746-67. doi: 10.1021/acs.jmedchem.6b01254 [Crossref] [ Google Scholar]